复方桔梗麻黄碱糖浆(II)的质量控制方法与流程
本发明属于药品质量控制
技术领域:
,尤其涉及一种复方桔梗麻黄碱糖浆(ii)的质量控制方法。
背景技术:
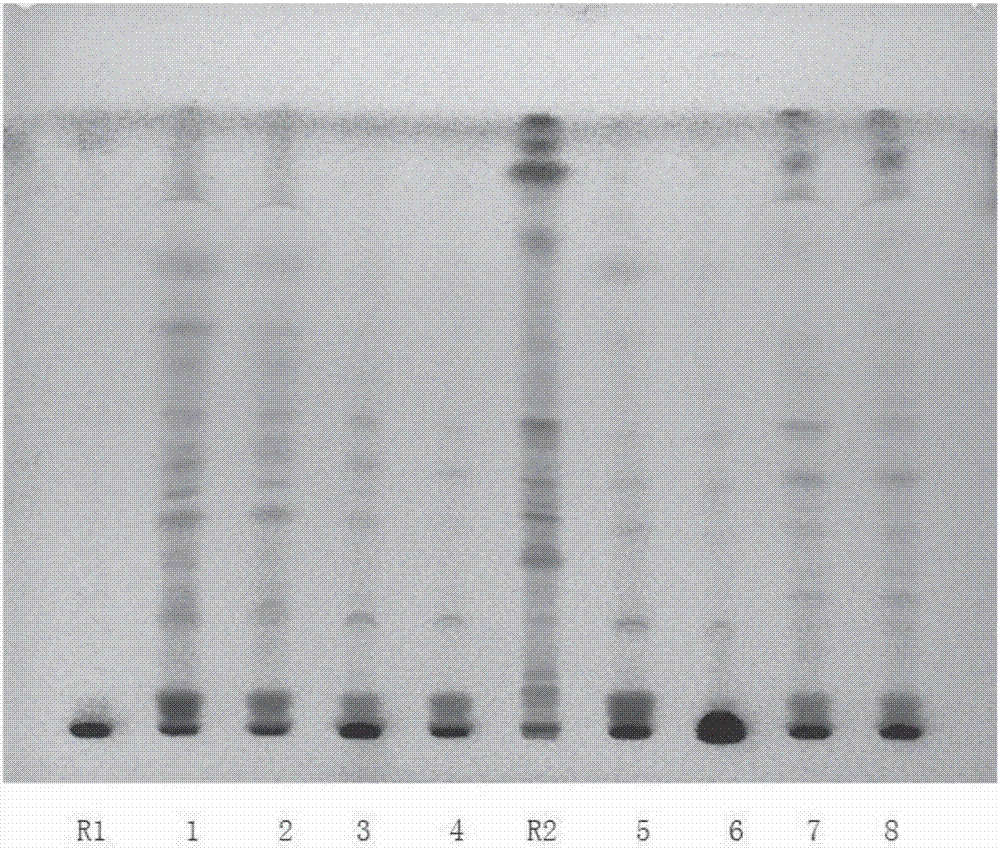
:复方桔梗麻黄碱糖浆(ii)(ii方)曾用名有小儿止咳糖浆、小儿咳喘糖浆、小儿咳嗽糖浆、小儿镇咳糖浆、小儿咳停糖浆、儿咳停等,现行标准为《国家药品标准》化学药品地方标准上升国家标准第十四册。成都世纪华洋制药有限责任公司1981年首先获得批文在国内上市。1982年即有多家企业获得批准文号。目前国内生产企业共有7家,7个批准文号。复方桔梗麻黄碱糖浆(ii)的现行标准检测项目设置有缺漏,对桔梗流浸膏、茴香水等中药提取物均无相关制法描述或要求;缺少对桔梗流浸膏的鉴别及含量控制项,对薄荷脑、蔗糖、防腐剂的含量也没有具体要求;盐酸麻黄碱的测定方法为uv法,前处理方法繁琐,方法专属性差。为此,现有方法难以有效控制生产中的成品质量,不同批次产品的质量没有合理准确的指标来进行衡量,无法确保产品疗效,对临床治疗影响巨大。技术实现要素:本发明要解决的技术问题是提供一种设计合理、准确可靠、专属性强且操作简单的复方桔梗麻黄碱糖浆(ii)的质量控制方法,以帮助有效控制复方桔梗麻黄碱糖浆(ii)生产、流通及使用等环节中的药品质量和提高其质量水平,确保临床使用疗效。为解决上述技术问题,本发明采用以下技术方案:复方桔梗麻黄碱糖浆(ii)的质量控制方法,包括以下步骤:<1>盐酸麻黄碱的薄层鉴别;<2>桔梗的薄层鉴别;<3>薄荷脑的顶空毛细管气相色谱法含量测定;<4>乙醇的顶空毛细管气相色谱法含量测定;<5>防腐剂的hplc法含量测定;<6>盐酸麻黄碱的hplc法含量测定;<7>氯化铵的定氮法含量测定。步骤<1>按以下操作进行:取本品5ml,加氢氧化钠试液使成碱性,加乙醚15ml振摇提取10分钟,分取醚层,挥去乙醚浓缩至约1ml作为供试品溶液;另取盐酸麻黄碱对照品,用乙醇制成每1ml中含盐酸麻黄碱500μg的溶液作为对照品溶液;照薄层色谱法试验,吸取上述溶液各10μl,分别点于同一硅胶g薄层板上,以正丁醇-醋酸-水(4∶1∶5)的上层溶液为展开剂,展开,晾干,喷以0.2%茚三酮的丙酮溶液,于105℃加热1~2分钟。步骤<2>按以下操作进行:取本品60ml,用乙酸乙酯振摇提取3次,每次60ml,静置分层,合并乙酸乙酯层,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取桔梗对照药材1g,加甲醇50ml,超声提取30min,滤过,取滤液蒸干,残渣加水60ml使溶解,自“用乙酸乙酯振摇提取3次”起,同法制成桔梗对照药材溶液;照薄层色谱法试验,吸取上述溶液各10μl,分别点于同一硅胶g薄层板上,以氯仿-甲醇-水(16∶4∶1)的下层溶液为展开剂,展开,晾干,喷以10%香草醛硫酸溶液,于105℃加热至斑点显色清晰。步骤<3>按以下操作进行:精密量取本品5ml置于20ml顶空瓶中,密封,作为供试品溶液;另取薄荷脑对照品约15mg置于100ml量瓶中,精密加入乙醇1.5ml使溶解后,加水稀释至刻度,摇匀,精密量取5ml置于20ml顶空瓶中,密封,作为混合对照品溶液;照气相色谱法测定;以6%氰丙基苯基-94%二甲基聚硅氧烷为固定液的毛细管柱为色谱柱;起始柱温为90℃,维持2分钟,以每分钟3℃的速率升温至140℃,维持6分钟,再以每分钟7℃的速率升温至200℃,维持5分钟;进样口温度为200℃;检测器温度为250℃;顶空瓶平衡温度为90℃,平衡时间为20分钟;取供试品溶液与混合对照品溶液分别顶空进样,记录色谱图。步骤<4>按以下操作进行:照步骤<3>薄荷脑项下的方法检查。步骤<5>按照高效液相色谱法测定;色谱条件与系统适用性溶液用十八烷基硅烷键合硅胶为填充剂:以0.02mol/l醋酸铵溶液为流动相a,甲醇为流动相b,按下表进行梯度洗脱,检测波长为255nm;取对照品溶液20μl注入液相色谱仪,记录色谱图;表1梯度洗脱流动相测定法精密称取本品约2g置50ml量瓶中,用50%甲醇溶液稀释至刻度,作为供试品溶液;精密称取苯甲酸、山梨酸、对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯对照品适量,用50%甲醇溶液溶解并稀释制成每1ml中含苯甲酸约60μg,含山梨酸、对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯约20μg的溶液,作为对照品溶液;精密量取上述溶液各20μ1注入液相色谱仪,记录色谱图。步骤<6>按照高效液相色谱法测定;色谱条件与系统适用性溶液用十八烷基硅烷键合硅胶为填充剂:以0.1%磷酸溶液为流动相a,甲醇为流动相b,按下表进行梯度洗脱,检测波长为205nm;取盐酸麻黄碱对照品和盐酸伪麻黄碱对照品适量,用0.1%磷酸溶液-甲醇(90∶10)溶解并稀释制成每1ml中约含盐酸麻黄碱16μg和盐酸伪麻黄碱1.6μg的混合溶液,作为系统适用性溶液,取20μl系统适用性溶液注入液相色谱仪,记录色谱图;表2梯度洗脱流动相时间min流动相a流动相b090%10%1290%10%1710%90%2390%10%2890%10%测定法精密称取本品2g置100ml量瓶中,用0.1%磷酸溶液-甲醇(90∶10)稀释至刻度,作为供试品溶液;另取盐酸麻黄碱对照品适量,用0.1%磷酸溶液-甲醇(90∶10)溶解并稀释成每1ml中约含盐酸麻黄碱16μg的溶液,作为对照品溶液;精密量取上述溶液各20μl注入液相色谱仪,记录色谱图;按相对密度折算,用外标法以峰面积计算,即得。步骤<7>按以下操作进行:取本品约5.8g,精密称定,照氮测定法依法测定。针对现有复方桔梗麻黄碱糖浆(ii)有关制剂质量检测方法存在的问题,发明人进行了改进,主要是增加了盐酸麻黄碱、桔梗的薄层鉴别,以及采用顶空毛细管气相色谱法进行薄荷脑和乙醇含量测定、采用hplc法进行盐酸麻黄碱含量测定、采用定氮法进行氯化铵含量测定。本发明的质量控制方法设计合理、准确可靠、专属性强且操作简单,据此建立复方桔梗麻黄碱糖浆(ii)新的质量控制标准,可帮助有效控制复方桔梗麻黄碱糖浆(ii)生产、流通及使用等环节中的药品质量和提高其质量水平,保证生产工艺可控,确保临床使用安全,为产品疗效稳定确切提供了可靠保障。附图说明图1是盐酸麻黄碱薄层图谱,图中:r1-阴性,r2-对照品,1~6-供试品溶液1~6。图2是桔梗薄层图谱,图中:r1-桔梗阴性,r2-桔梗对照药材,1~8-供试品溶液1~8。图3是阴性空白溶液gc色谱图。图4是对照品溶液gc色谱图。图5是供试品gc色谱图。图6是干扰因素(茴香水溶液)gc色谱图。图7是乙醇对照溶液gc色谱图。图8是供试品gc色谱图。图9是阴性空白溶液gc色谱图。图10是干扰因素(茴香水溶液)gc色谱图。图11是阴性样品溶液hplc图。图12是对照品溶液hplc图,图中:1.苯甲酸;2.山梨酸;3.对羟基苯甲酸甲酯4.对羟基苯甲酸乙酯5.对羟基苯甲酸丙酯6.对羟基苯甲酸丁酯。图13是供试品溶液hplc图。图14是对照品溶液hplc图。图15是供试品溶液hplc图。图16是阴性样品溶液hplc图。具体实施方式复方桔梗麻黄碱糖浆(ii)的质量控制方法研究<1>盐酸麻黄碱的薄层鉴别拟定方法如下:取本品5ml,加氢氧化钠试液使成碱性,加乙醚15ml振摇提取10分钟,分取醚层,挥去乙醚浓缩至约1ml作为供试品溶液;另取盐酸麻黄碱对照品,用乙醇制成每1ml中含盐酸麻黄碱500μg的溶液作为对照品溶液;照薄层色谱法(中国药典通则0502)试验,吸取上述溶液各10μl,分别点于同一硅胶g薄层板上,以正丁醇-醋酸-水(4∶1∶5)的上层溶液为展开剂,展开,晾干,喷以0.2%茚三酮的丙酮溶液,于105℃加热1~2分钟;供试品所显示斑点的颜色和位置应与对照品相同。按拟定方法鉴别6批次样品,结果如图1。在供试品色谱中,与盐酸麻黄碱对照品在相同位置上均显相同颜色的斑点,按处方工艺制备的盐酸麻黄碱阴性样品无干扰,验证方法可行。<2>桔梗的薄层鉴别拟定方法如下:取本品60ml,用乙酸乙酯振摇提取3次,每次60ml,静置分层,合并乙酸乙酯层,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取桔梗对照药材1g,加甲醇50ml,超声提取30min,滤过,取滤液蒸干,残渣加水60ml使溶解,自“用乙酸乙酯振摇提取3次”起,同法制成桔梗对照药材溶液;照薄层色谱法(中国药典通则0502)试验,吸取上述溶液各10μl,分别点于同一硅胶g薄层板上,以氯仿-甲醇-水(16∶4∶1)的下层溶液为展开剂,展开,晾干,喷以10%香草醛硫酸溶液,于105℃加热至斑点显色清晰,供试品色谱中,在与对照药材色谱相应的位置上,显2~3个相同颜色的斑点。按拟定方法鉴别8批次样品,结果如图2。在供试品色谱中,与桔梗对照药材在相同位置上均显相同颜色的斑点,按处方工艺制备的桔梗阴性样品无干扰,验证方法可行。<3>薄荷脑的顶空毛细管气相色谱法含量测定拟定方法如下:精密量取本品5ml置于20ml顶空瓶中,密封,作为供试品溶液;另取薄荷脑对照品约15mg置于100ml量瓶中,精密加入乙醇1.5ml使溶解后,加水稀释至刻度,摇匀,精密量取5ml置于20ml顶空瓶中,密封,作为混合对照品溶液;照气相色谱法(中国药典通则0521)测定;以6%氰丙基苯基-94%二甲基聚硅氧烷(或极性相似)为固定液的毛细管柱为色谱柱;起始柱温为90℃,维持2分钟,以每分钟3℃的速率升温至140℃,维持6分钟,再以每分钟7℃的速率升温至200℃,维持5分钟;进样口温度为200℃;检测器温度为250℃;顶空瓶平衡温度为90℃,平衡时间为20分钟;取供试品溶液与混合对照品溶液分别顶空进样,记录色谱图,按外标法以峰面积计算,薄荷脑含量应不低于75%。经试验验证样品中其他挥发性成分对薄荷脑的测定均无干扰(见图3-6),方法进行了专属性考察、线性关系考察、进样精密度、检测限及限度确定等方法学研究工作,验证方法可行。其中,线性回归分析,结果显示,薄荷脑在浓度为81.34μg/ml~1779μg/ml的范围内,线性关系成立,线性方程为y=3494278x+153918,r=0.9995。加样回收试验平均回收率为92.57%及rsd值为7.1%。精密度试验结果良好,rsd为5.4%。检出限(s/n=3)与定量限(s/n=10)结果分别为0.0943μg,0.314μg。<4>乙醇的顶空毛细管气相色谱法含量测定拟定方法如下:照步骤<3>薄荷脑项下的方法检查,乙醇量应为1.35%~3.22%(ml/ml)。经试验验证样品中其他挥发性成分对乙醇的测定均无干扰(见图7-10),方法进行了专属性考察、线性关系考察、进样精密度、检测限及限度确定等方法学研究工作,验证方法可行。其中,线性回归分析,结果显示,乙醇在浓度为0.006679%~0.03817%(ml/ml)的范围内,线性关系成立,线性方程为y=1618386363x+753625,r=0.9996。加样回收试验平均回收率为97.74%及rsd值为1.63%。精密度试验结果良好,rsd为2.3%。检出限(s/n=3)与定量限(s/n=10)结果分别为0.0018%(ml/ml),0.0061%(ml/ml)。<5>防腐剂的hplc法含量测定拟定方法如下:按照高效液相色谱法(中国药典通则0512)测定;色谱条件与系统适用性溶液用十八烷基硅烷键合硅胶为填充剂:以0.02mol/l醋酸铵溶液(冰醋酸调ph至4.0)为流动相a,甲醇为流动相b,按下表进行梯度洗脱,检测波长为255nm;取对照品溶液20μl注入液相色谱仪,记录色谱图,各防腐剂之间分离度应符合规定;表3梯度流动相时间(min)流动相a流动相b080%20%555%45%3055%45%3580%20%4580%20%测定法精密称取本品约2g置50ml量瓶中,用50%甲醇溶液稀释至刻度,作为供试品溶液;精密称取苯甲酸、山梨酸、对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯对照品适量,用50%甲醇溶液溶解并稀释制成每1ml中含苯甲酸约60μg,含山梨酸、对羟基苯甲酸甲酯、对羟基苯甲酸乙酯、对羟基苯甲酸丙酯、对羟基苯甲酸丁酯约20μg的溶液,作为对照品溶液;精密量取上述溶液各20μl注入液相色谱仪,记录色谱图;按相对密度折算,用外标法以峰面积计算供试品中防腐剂含量,其中苯甲酸含量应为标示量的80%~120%,其他防腐剂应不得检出。发明人进行了流动相考察、检测波长的选择、溶剂选择、专属性考察、线性关系考察、溶液稳定性、进样精密度、检测限及限度确定等方法学研究工作,证明拟定方法可行(图11-13)。dad检测器扫描结果显示,苯甲酸、山梨酸的吸收波长分别为231nm、258nm、,对羟基苯甲酸酯类的吸收波长均为255nm,盐酸麻黄碱的吸收波长为205nm,因此我们选择255nm作为苯甲酸、山梨酸、对羟基苯甲酸酯类的测定波长。各防腐剂线性关系考察显示,在相应的浓度范围内线性关系均良好(见下表)。表4各防腐剂线性关系考察结果名称回归方程相关系数浓度范围μg/ml苯甲酸a=5456.55c+17289.920.9997290.2500-0.5805山梨酸a=186643.52c+25297.330.999958.2000-0.5820对羟基苯甲酸甲酯a=44142.52c+31893.020.9997108.2652-0.2707对羟基苯甲酸乙酯a=40586.60c+34360.070.9997118.6812-0.2967对羟基苯甲甲酸丙酯a=40353.98c+15785.530.9999111.9000-0.2798对羟基苯甲酸丁酯a=33085.67c+33228.840.9996127.4000-0.3185进样精密度考察与稳定性考察12小时结果良好,见下表。表5进样精密度考察与稳定性考察结果名称进样精密度考察rsd(%)稳定性考察rsd(%)苯甲酸0.21.0山梨酸0.10.1对羟基苯甲酸甲酯0.10.2对羟基苯甲酸乙酯0.10.2对羟基苯甲酸丙酯0.71.8对羟基苯甲酸丁酯0.30.2各防腐剂定量限(s/n=10)和检测限(s/n=3)考察结果见表6。表6各防腐剂定量限和检测限考察结果名称检出限(g/kg)定量限(g/kg)苯甲酸0.00260.0087山梨酸0.000100.00035对羟基苯甲酸甲酯0.000530.0018对羟基苯甲酸乙酯0.000830.0028对羟基苯甲酸丙酯0.00140.0045对羟基苯甲酸丁酯0.00240.0081<6>盐酸麻黄碱的hplc法含量测定拟定方法如下:按照高效液相色谱法(中国药典通则0512)测定;色谱条件与系统适用性溶液用十八烷基硅烷键合硅胶为填充剂:以0.1%磷酸溶液(v/v,用三乙胺调ph至3.0)为流动相a,甲醇为流动相b,按下表进行梯度洗脱,检测波长为205nm;取盐酸麻黄碱对照品和盐酸伪麻黄碱对照品适量,用0.1%磷酸溶液(v/v,用三乙胺调ph至3.0)-甲醇(90∶10)溶解并稀释制成每1ml中约含盐酸麻黄碱16μg和盐酸伪麻黄碱1.6μg的混合溶液,作为系统适用性溶液,取20μl系统适用性溶液注入液相色谱仪,记录色谱图,盐酸麻黄碱峰与盐酸伪麻黄碱峰的分离度应符合规定;表7梯度流动相时间(min)流动相a流动相b090%10%1290%10%1710%90%2390%10%2890%10%测定法精密称取本品2g置100ml量瓶中,用0.1%磷酸溶液(v/v,用三乙胺调ph至3.0)-甲醇(90:10)稀释至刻度,作为供试品溶液;另取盐酸麻黄碱对照品适量,用0.1%磷酸溶液(v/v,用三乙胺调ph至3.0)-甲醇(90∶10)溶解并稀释成每1ml中约含盐酸麻黄碱16μg的溶液,作为对照品溶液;精密量取上述溶液各20μl注入液相色谱仪,记录色谱图;按相对密度折算,用外标法以峰面积计算,即得。发明人进行了流动相考察、检测波长的选择、溶剂选择、专属性考察、线性关系考察、溶液稳定性、进样精密度、检测限及限度确定等方法学研究工作,证明拟定方法可有效分离盐酸麻黄碱(图14-16)。其中,线性考察得回归方程a=46.9947c-9.2234(r=0.9999)。结果表明盐酸麻黄碱在1.8115-90.5775μg/ml的浓度范围内线性关系良好。回收率考察中制得高、中、低三个浓度(相当于测定浓度80%、100%、120%)的溶液共9份按拟定色谱条件分别进样,结果见表8。表8回收率考察结果取线性关系考察项下线性溶液重复进样5次,结果rsd为0.5%。表明仪器和稳定性符合要求。取线性关系考察项下线性溶液在24个小时内每隔3小时进样一次。结果rsd为0.3%。表明溶液在24小时内稳定。定量限和检测限考察中,用信噪比(s/n)为3及10来确定各成分的定量限与检测限,结果检测限为0.0033g/kg,定量限为0.11g/kg。<7>氯化铵的定氮法含量测定拟定方法如下:取本品约5.8g,精密称定,照氮测定法(中国药典通则0704第三法)依法测定;按相对密度折算,每1ml硫酸滴定液(0.05mol/l)相当于5.349mg的nh4cl。为考察制剂中辅料对氯化铵测定的影响,同样按处方工艺配制不含氯化铵的阴性样品。实验结果显示,连续测定20份氯化铵的阴性样品,消耗的硫酸滴定液(0.05mol/l)按氯化铵折算,都在0.15mg/ml~0.20mg/ml之间,按氯化铵标示量(15mg/ml)折算约为标示含量的1.2%左右,即样品中的辅料对氯化铵测定结果基本无干扰,可忽略不计,方法可行。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1