大黄甘草汤的质量控制方法与流程
本发明涉及一种中药材的质量控制方法,具体涉及大黄甘草汤的质量控制方法。
背景技术
大黄和甘草药对是医家常用泻下药对。大黄苦寒,清泄胃中结热,力猛善行,甘草甘平,与大黄相伍,可缓大黄之泻下,留大黄于胃以洁府,免大黄苦寒伤中,使其调中有补以愈疾。两药配伍,相须为用,甘草缓和大黄苦寒攻下之性,使其降逆止呕而不伤胃气,又可清泄胃热,大黄甘草汤也是由这两味中药组成。
大黄含有多种蒽醌类成分,甘草中含有多个皂苷类成分,目前的检测方法,检测的有效成分比较有限,不能全面检测大黄和甘草药对中的有效成分。因此,为了充分控制好大黄和甘草药对及其制剂的临床安全性,维护患者的利益,很有必要在现有技术的基础之上研究设计出能准确全面检测大黄甘草汤及其制剂有效成分的检测方法。
技术实现要素:
发明目的:本发明的目的是解决现有技术的不足,经过大量实验筛选,采用超高效液相色谱检测,该检测方法可以同时检测大黄甘草汤中的蒽醌类、甾体类共17种活性化合物。该方法检测灵敏度高,稳定性好,可以客观、全面、准确地评价大黄甘草汤及其制剂的质量,对控制质量和保证疗效具有重要意义。
技术方案:为了实现以上目的,本发明采取的技术方案为:
一种大黄甘草汤的质量控制方法,包括以下步骤:
(1)样品溶液的制备
先取甘草药材,加8至16倍量的水回流提取,然后加入大黄药材,并加入药材总重量8~16倍的水,回流提取,过滤得到大黄甘草水提液,离心,吸取上清液,过0.22μm滤膜,得样品溶液;
(2)对照品溶液的制备
精密称定大黄酸、大黄素、芦荟大黄素、大黄酚、大黄素甲醚、大黄酸-8-o-β-d-葡萄糖苷、大黄素-8-o-β-d-葡萄糖苷、番泻苷a、番泻苷b、甘草酸、甘草次酸、甘草苷、异甘草苷、甘草素、异甘草素、甘草查耳酮a和光甘草定17种对照品各,分别用甲醇溶解得到对照品储备液,然后定量吸取各对照品储备液获得混合对照品储备液,过0.22μm滤膜,得混合对照品溶液;
(4)定量分析
将步骤(1)制备得到的样品溶液和步骤(2)制备得到的混合对照品溶液,分别进行超高效液相色谱检测,分别获得样品和对照品的uhplc-tq-ms/ms色谱图,然后进行定性定量分析。
作为优选方案,以上所述的大黄甘草汤的质量控制方法,步骤(3)所用超高效液相色谱参数设置如下:
仪器:watersacquityuplc系统;tetons质谱检测器;
色谱柱:thermoscientifichypersilgoldc18色谱柱,100mm×2.1mm,1.9μm,;柱温:35℃;进样体积:2μl;流动相:a相为0.1%甲酸水、b相为乙腈;流速:0.4ml·min-1;梯度洗脱;
质谱条件为:离子源:esi离子源;离子源温度:150℃;扫描方式:多反应检测模式;毛细管电压:3.0kv;去溶剂气温度:150℃;去溶剂气流量:1000l·h-1;锥孔气流量:30l·h-1;碰撞气流量:0.15ml·min-1。
作为优选方案,以上所述的大黄甘草汤的质量控制方法,梯度洗脱程序为:0~2min,a:95%~95%,b:5%~5%;2~4min,a:95%~65%,b:5%~35%;4~8min,a:65%~5%,b:35%~95%;8~13min,a:5%~5%,b:95%~95%;13~14min,a:5%~95%,b:95%~5%;14~15min,a:95%~95%,b:5%~5%。
作为优选方案,以上所述的大黄甘草汤的质量控制方法,步骤(3)中用90%的甲醇按照逐级稀释法对混合对照品溶液进行逐级稀释,制成5个不同浓度级别的对照品溶液分别进行超高效液相色谱检测,测定各对照品溶液中17个成分的峰面积,以各成分浓度作为横坐标,峰面积为纵坐标绘制17个成分的标准曲线,采用回归法进行线性分析,计算各对照品的线性相关系数r2;并测定loq和lod,如下表
作为优选方案,以上所述的大黄甘草汤的质量控制方法,步骤(2)对照品溶液的制备方法为:
精密称定大黄酸、大黄素、芦荟大黄素、大黄酚、大黄素甲醚、大黄酸-8-o-β-d-葡萄糖苷、大黄素-8-o-β-d-葡萄糖苷、番泻苷a、番泻苷b、甘草酸、甘草次酸、甘草苷、异甘草苷、甘草素、异甘草素、甘草查耳酮a和光甘草定对照品各5mg左右,用90%甲醇溶解为浓度分别为大黄酸108.6μg·ml-1、大黄素,106.0μg·ml-1、芦荟大黄素220.0μg·ml-1、大黄酚90.0μg·ml-1、大黄素甲醚130.0μg·ml-1、大黄酸-8-o-β-d-葡萄糖苷1347.0μg·ml-1、大黄素-8-o-β-d-葡萄糖苷408.3μg·ml-1、番泻苷a126.0μg·ml-1、番泻苷b245.0μg·ml-1、甘草酸1353.0μg·ml-1、甘草次酸721.4μg·ml-1、甘草苷505.7μg·ml-1、异甘草苷3320.0μg·ml-1、甘草素3233.0μg·ml-1、异甘草素3553.0μg·ml-1、甘草查耳酮2127.0μg·ml-1、光甘草定2647.0μg·ml-1的对照品储备液;然后定量吸取各对照品储备液获得混合对照品储备液,其中大黄酸(1)、大黄素(2)、芦荟大黄素(3)、大黄酚(4)、大黄素甲醚(5)、大黄酸-8-o-β-d-葡萄糖苷(6)、大黄素-8-o-β-d-葡萄糖苷(7)、番泻苷a(8)、番泻苷b(9)、甘草酸(10)、甘草次酸(11)、甘草苷(12)、异甘草苷(13)、甘草素(14)、异甘草素(15)、甘草查耳酮a(16)、光甘草定(17)的含量分别为15.404μg·ml-1、15.035μg·ml-1、15.603μg·ml-1、12.766μg·ml-1、18.440μg·ml-1、19.106μg·ml-1、14.479μg·ml-1、17.872μg·ml-1、17.376μg·ml-1、19.191μg·ml-1、15.349μg·ml-1、14.346μg·ml-1、23.546μg·ml-1、22.929μg·ml-1、25.199μg·ml-1、15.085μg·ml-1、23.005μg·ml-1。
作为优选方案,以上所述的大黄甘草汤的质量控制方法,步骤(1)所述的样品溶液的制备方法为:
先取甘草药材,加8倍量的水回流提取1h后,然后加入大黄药材,并加入药材总重量8倍的水,回流提取2次,合并提取液,过滤得到大黄甘草水提液,离心,吸取上清液,过0.22μm滤膜,得样品溶液。大黄和甘草的重量比为4:1、4:2、4:3、4:4、3:4、2:4或1:4。
条件优化实验
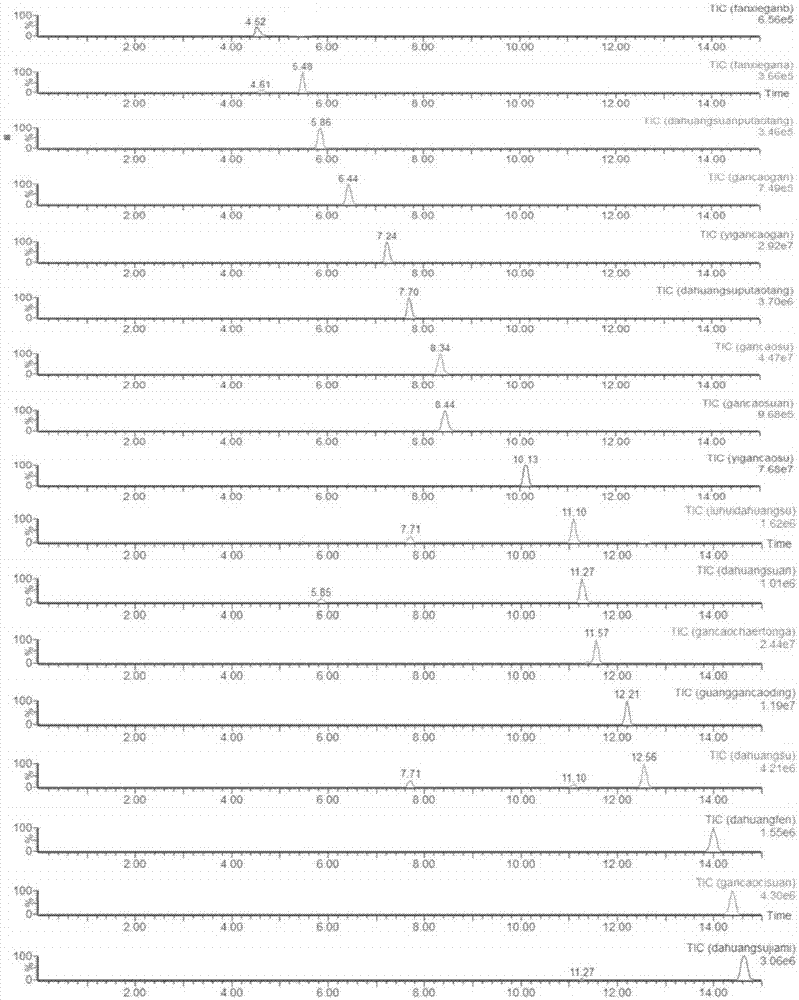
本发明通过对流动相、梯度洗脱程序、流速等条件进行考察,以期所有成分能够在较短时间内即能分离又能获得较好的峰形。发现以0.1%的甲酸水-乙腈能够很好的分离大黄-甘草中的17种活性成分,加入甲酸能够减少芳香酸类成分的拖尾现象;通过优化梯度洗脱程序发现高比例有机相对大黄-甘草中活性成分分离较好,确定洗脱梯度;流动相流速为0.4ml·min-1各成分分离效果及峰形优于流速为0.2ml·min-1。采用全扫模式(正离子及负离子模式)优化17种化合物的质谱参数,仪器在分析各成分在正离子及负离子模式下的信号强度及灵敏度选择检测模式,锥孔电压及毛细管电压由软件自动优化。在色谱及质谱条件优化后获得大黄-甘草中17种活性成分的uhplc-tq-ms/ms色谱图如图1所示。
有益效果:本发明提供的大黄甘草汤的质量控制方法和现有技术相比具有以下优点:
本发明根据大黄甘草汤中蒽醌和皂苷2类不同成分,共17种不同结构和性质的活性化合物的结构及其性质特点,通过大量实验筛选出最佳的流动相组成,洗脱程序、流速,色谱柱、检测器检测参数等分析条件。经多次实验验证表明,本发明能够同时检测蒽醌和皂苷2类不同成分共17种化合物,该方法检测灵敏度高,稳定性好,可以客观、全面、准确的评价大黄甘草汤及其制剂的质量,对控制质量和保证临床疗效具有重要意义。
附图说明
图1为大黄甘草汤中17个成分的uhplc-tq-ms/ms的多反应检测模式(mrm)色谱图。
具体实施方式
下面结合具体实施例进一步阐明本发明,应理解这些实施例仅用于说明本发明而不用于限制本发明的范围,在阅读了本发明之后,本领域技术人员对本发明的各种等价形式的修改均落于本申请所附权利要求所限定的范围。
实施例1
1、一种大黄甘草汤的质量控制方法,其包括以下步骤:
仪器与试剂
1.1仪器
watersacquityuhplc系统,购自美国waters公司;tetons质谱检测器,购自美国waters公司;masslynxtm质谱工作站软件,购自美国waters公司;超纯水仪,购自南京易普易达科技发展有限公司;micofuge22rcentrifuge型台式离心机,购自北京博雅创新科技发展有限公司;超声波清洗仪,购自昆山禾创超声仪器有限公司;
1.2药物与试剂
大黄(产地:甘肃,批号:20160319)、甘草(产地:宁夏,批号:151108),大黄为唐古特大黄,甘草为乌拉尔甘草,且符合药典标准。
化学标准品大黄酸(1)、大黄素(2)、芦荟大黄素(3)、大黄酚(4)、大黄素甲醚(5)、大黄酸-8-o-β-d-葡萄糖苷(6)、大黄素-8-o-β-d-葡萄糖苷(7)、番泻苷a(8)、番泻苷b(9)、甘草酸(10)、甘草次酸(11)、甘草苷(12)、异甘草苷(13)、甘草素(14)、异甘草素(15)、甘草查耳酮a(16)、光甘草定(17)购自成都瑞芬思生物科技有限公司,纯度大于98%。甲醇,色谱纯,购自德国merck试剂公司;乙腈,色谱纯,购自德国merck试剂公司。
2实验方法
2.1对照品溶液的制备
精密称定上述17种对照品各5mg左右,用90%甲醇溶解为浓度分别为大黄酸(1,108.6μg·ml-1)、大黄素(2,106.0μg·ml-1)、芦荟大黄素(3,220.0μg·ml-1)、大黄酚(4,90.0μg·ml-1)、大黄素甲醚(5,130.0μg·ml-1)、大黄酸-8-o-β-d-葡萄糖苷(6,1347.0μg·ml-1)、大黄素-8-o-β-d-葡萄糖苷(7,408.3μg·ml-1)、番泻苷a(8,126.0μg·ml-1)、番泻苷b(9,245.0μg·ml-1)、甘草酸(10,1353.0μg·ml-1)、甘草次酸(11,721.4μg·ml-1)、甘草苷(12,505.7μg·ml-1)、异甘草苷(13,3320.0μg·ml-1)、甘草素(14,3233.0μg·ml-1)、异甘草素(15,3553.0μg·ml-1)、甘草查耳酮(16,2127.0μg·ml-1)、光甘草定(17,2647.0μg·ml-1)的对照品储备液。定量吸取各对照品储备液获得混合对照品储备液,其中大黄酸(1)、大黄素(2)、芦荟大黄素(3)、大黄酚(4)、大黄素甲醚(5)、大黄酸-8-o-β-d-葡萄糖苷(6)、大黄素-8-o-β-d-葡萄糖苷(7)、番泻苷a(8)、番泻苷b(9)、甘草酸(10)、甘草次酸(11)、甘草苷(12)、异甘草苷(13)、甘草素(14)、异甘草素(15)、甘草查耳酮a(16)、光甘草定(17)的含量分别为15.404μg·ml-1、15.035μg·ml-1、15.603μg·ml-1、12.766μg·ml-1、18.440μg·ml-1、19.106μg·ml-1、14.479μg·ml-1、17.872μg·ml-1、17.376μg·ml-1、19.191μg·ml-1、15.349μg·ml-1、14.346μg·ml-1、23.546μg·ml-1、22.929μg·ml-1、25.199μg·ml-1、15.085μg·ml-1、23.005μg·ml-1。
2.2供试品溶液的制备
先取甘草药材,加8倍量的水回流提取1h后,然后加入大黄药材,并加入药材总重量8倍的水,回流提取2次,合并提取液,过滤得到大黄甘草水提液,离心,吸取上清液,过0.22μm滤膜,得样品溶液。大黄和甘草的重量比为4:1。
2.3色谱分析条件
色谱柱:thermoscientifichypersilgoldc18色谱柱(100mm×2.1mm,1.9μm);柱温:35℃;进样体积:2μl;流动相:a(0.1%甲酸水)、b(乙腈);流速:0.4ml·min-1;梯度洗脱:0~1min,a:95%~95%,b:5%~5%;2~4min,a:95%~65%,b:5%~35%;4~8min,a:65%~5%,b:35%~95%;8~13min,a:5%~5%,b:95%~95%;13~14min,a:5%~95%,b:95%~5%;14~15min,a:95%~95%,b:5%~5%。
2.4质谱条件
离子源:esi离子源;离子源温度:150℃;扫描方式:多反应检测模式(mrm);毛细管电压:3.0kv;去溶剂气温度:150℃;去溶剂气流量:1000l·h-1;锥孔气流量:30l·h-1;碰撞气流量:0.15ml·min-1。各成分保留时间及质谱参数见表1。
表1各成分保留时间及质谱参数
2.5方法学考察
2.5.1线性关系、定量限(lod)、检测限(loq)
用90%的甲醇按照逐级稀释法对对照品储备液进行逐级稀释,制成不同浓度级别的对照品溶液按照2.3及2.4项下的色谱及质谱检测方法测定各对照品溶液中17个成分的峰面积,以各成分浓度作为横坐标,峰面积为纵坐标绘制这17个成分的标准曲线,采用回归法进行线性分析,计算各对照品的线性相关系数(r2);信噪比为3、10时的对照品浓度分别为loq、lod。如下表2:
表2各成分标准曲线和线性范围和loq、lod
2.5.2精密度、重复性、稳定性
精密度考察:将一份对照品溶液按照以上色谱及质谱检测方法测定各对照品溶液中17个成分的峰面积,每天重复进样3次,连续3天及同一天内连续进样6次,用各成分峰面积的相对标准偏差(rsd)评价样品日间及日内精密度。
重复性考察:平行制备6份大黄-甘草1:1配比的供试品溶液,按上述色谱及质谱检测方法测定供试样品中各成分的峰面积,用各成分峰面积的相对标准偏差(rsd)评价来评价仪器的重复性。
稳定性考察:取大黄-甘草1:1配比的供试品溶液在1、2、4、8、12、24h时分别进样,测定不同时间供试样品中17种化合物的含量变化,以这17种化合物峰面积的rsd值评价样品的稳定性。
2.5.3回收率实验
在已知17个成分含量的样品中加入其含量80%、100%、120%的对照品溶液,供试品溶液按上述色谱及质谱检测方法测定样品峰面积,根据标准曲线获得样品浓度,计算加样回收率,加样回收率=(加标试样浓度-试样浓度)/加标量。结果表明大黄及甘草中17种活性成分回收率在98.16%~100.64%范围内,准确性好。
2.6统计学分析
所有结果均运用spss22.0软件进行处理,采用anova中dunnett法进行统计学差异分析,p<0.05表示差异具有统计学意义,所有结果采用平均值±标准差(mean±sd)表示。
3.实验结果
如表3,本发明精密度、重复性、稳定性,结果表明其rsd均小于3%,精密度好,大黄及甘草中17种活性成分回收率在98.16%~100.64%范围内。
表3精密度、重复性、稳定性和加样回收实验
本发明提供的检测方法能同时准确检测17个化合物,并且本发明提供的大黄甘草汤的质量控制方法,精密度高,灵敏度高,稳定性和准确度高,可以客观、全面、准确地评价大黄甘草汤的质量,对准确控制大黄甘草汤的质量具有重要意义。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!