检测维生素C药物中维生素C及异维生素C含量的方法与流程
本发明涉及药物分析
技术领域:
,尤其涉及一种采用高效液相色谱法检测维生素c药物中维生素c及异维生素c含量的方法,主要用于检测维生素c原料药及制剂中主药维生素c及杂质异维生素c的含量。
背景技术:
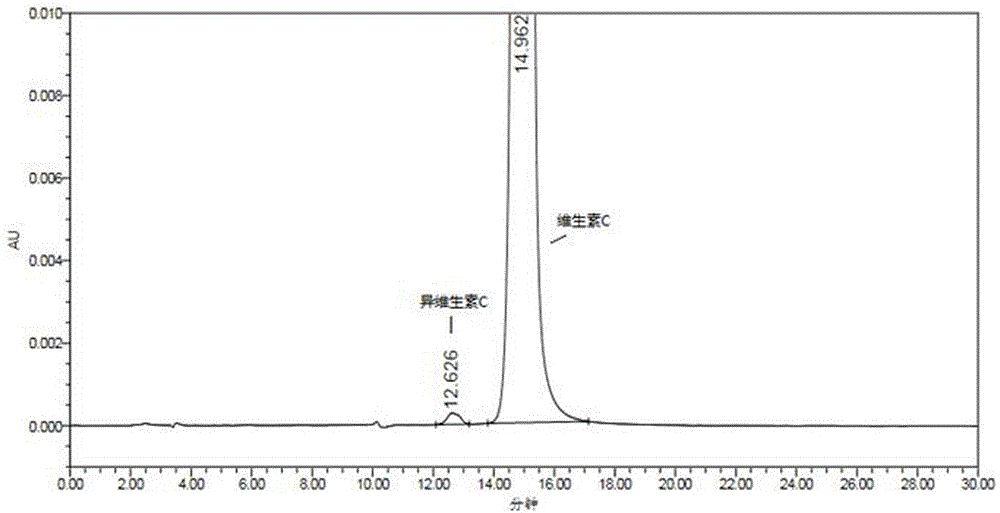
:维生素c药物中的主药为维生素c,维生素c的化学名称为l-坏血酸,分子式为c6h8o6,分子量为176.13,结构式为:维生素c参与氨基酸代谢、神经递质的合成、胶原蛋白和组织细胞间质的合成,可降低毛细血管的通透性,加速血液的凝固,刺激凝血功能,促进铁在肠内的吸收,促使血脂下降,增加对感染的抵抗力,参与解毒功能,且有抗组胺的作用及阻止致癌物质(亚硝胺)生成的作用。目前国内维生素c药物中维生素c含量的测定均采用容量滴定法进行测定,但维生素c中含有同分异构体(异维生素c),异维生素c结构式为:在滴定过程中异维生素c同样会消耗滴定液,导致检测结果不准确。另外,现有技术中还公开了专属性更强的高效液相色谱(hplc)法检测维生素c含量的方法,如中国专利公告号为cn103353499a的现有技术在2013年10月16日公开了一种恒流液相色谱法同时测定古龙酸、古龙酸甲酯和维生素c的方法。该方法采用恒流,对仪器设备要求低。该方法同时测定古龙酸、古龙酸甲酯和维生素c,避免了采用两种或以上方法测定同一样品中的不同组份,方法快速,整个分离过程不足10min,且三种组分的分离度均大于1.5,符合相关色谱法对分离度的要求。但该方法仍不能很好地检出测维生素c药物中维生素c含量,这是因为维生素c结构式中含有多个酸性极性基团,在普通的反相柱上保留较弱,与异维生素c分离不开。因此,有必要使用对极性物质选择性更高的色谱柱,开发出能使维生素c药物中的维生素c与异维生素c良好分离,并实现对维生素c含量及异维生素c含量准确检测的新技术。技术实现要素:本发明的目的在于解决现有技术中存在的上述问题,提供一种检测维生素c药物中维生素c及异维生素c含量的方法,本发明在检测过程中能使维生素c与其同分异构体异维生素c良好分离,并能够实现对维生素c药物中维生素c和异维生素c含量的准确检测。而且操作简便,容易控制,检测成本低,并具有良好的专属性、精密度,检测结果准确可靠,为监控维生素c药物中的主药含量及杂质含量提供了一种行之有效的检测方法,进一步保证了维生素c药物的品质和患者的用药安全。为实现上述目的,本发明采用的技术方案如下:一种检测维生素c药物中维生素c及异维生素c含量的方法,其特征在于:采用高效液相色谱法检测,检测条件如下:色谱柱:hilic色谱柱;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;检测器:紫外检测器;所述缓冲盐溶液为甲酸铵溶液、醋酸铵溶液中的一种或按任意比例混合的两种。所述混合液中缓冲盐溶液与乙腈的体积比为70—85:15—30。所述缓冲盐溶液的浓度为0.05—0.2mol/l。所述色谱柱的柱温为25—40℃。所述流动相的流速为0.8—1.2ml/min。所述检测器波长为210—280nm。所述缓冲盐溶液的ph值为5.5—7.0,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别取维生素c药物与异维生素c的对照品,加稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c0.3—30μg的混合溶液,作为系统适用性试验溶液,精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000;(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。所述稀释剂为乙腈与有机酸溶液按任意比例的混合。所述有机酸溶液为醋酸铵溶液或冰醋酸溶液,为醋酸铵溶液时,醋酸铵溶液的浓度为0.05mol/l,ph值为6.0;为冰醋酸溶液时,冰醋酸溶液的体积百分比浓度为0.3%。采用本发明的优点在于:本发明提供了一种检测维生素c药物中主药维生素c含量及杂质异维生素c含量的新方法,在此检测方法中,由于采用了亲水性的hilic色谱柱,hilic色谱柱是近年来色谱领域研究的热点之一,此类色谱柱对极性化合物有较高的选择性,维生素c与异维生素c由于空间构型上的不同,使得二者的极性稍有差异,在亲水性的hilic色谱柱上二者能完全分离开,因此本方法可排除异维生素c的干扰,实现对维生素c含量的准确检测,同时可实现对维生素c中异维生素c含量的准确检测。而且操作简便,容易控制,检测成本低,并具有良好的专属性、精密度,检测结果准确可靠,为监控维生素c药物中的主药含量提供了一种行之有效的检测方法,进一步保证了维生素c药物的品质和患者的用药安全。附图说明图1为实施例1中检测系统适用性试验溶液的色谱图;图2为实施例2中检测系统适用性试验溶液的色谱图;图3为实施例3中检测系统适用性试验溶液的色谱图;图4为实施例4中检测系统适用性试验溶液的色谱图;图5为实施例5中检测系统适用性试验溶液的色谱图;图6为实施例6中检测系统适用性试验溶液的色谱图;图7为实施例7中检测系统适用性试验溶液的色谱图;具体实施方式实施例1本实施例是最优实施例,具体为:一种检测维生素c药物中维生素c及异维生素c含量的方法,采用高效液相色谱法检测,高效液相色谱的检测条件如下:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:30℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:1.0ml/min;检测器:紫外检测器;检测波长:268nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为80:20,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.1mol/l,缓冲盐溶液的ph值为6.8,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c0.3μg的混合溶液,作为系统适用性试验溶液。其中,所述稀释剂为乙腈与有机酸溶液按任意比例的混合,优选乙腈与有机酸溶液的体积比为50:50。所述有机酸溶液为醋酸铵溶液或冰醋酸溶液,为醋酸铵溶液时,醋酸铵溶液的浓度为0.05mol/l,ph值为6.0;为冰醋酸溶液时,冰醋酸溶液的体积百分比浓度为0.3%,本实施例优选有机酸溶液为0.3%冰醋酸溶液。然后精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图1和下表1:表1峰#保留时间分离度理论塔板数高度(微伏)对称因子异维生素c12.626——43932731.05维生素c14.9623.383111837090.96结合异维生素c与维生素c对照品的定位保留时间,图1中保留时间为12.626分钟的色谱峰是异维生素c的色谱峰,保留时间为14.962分钟的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。实施例2一种检测维生素c药物中维生素c及异维生素c含量的方法,所述高效液相色谱的检测条件为:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:30℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:1.0ml/min;检测器:紫外检测器;检测波长:268nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为85:15,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.2mol/l,缓冲盐溶液的ph值为6.8,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加入与实施例1相同稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c30μg的混合溶液,作为系统适用性试验溶液。精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图2和下表2:表2峰#保留时间峰高分离度理论塔板数对称因子异维生素c26.57829455——81771.07维生素c34.600847356.49113891.09结合异维生素c与维生素c对照品的定位保留时间,图2中保留时间为34.600的色谱峰是异维生素c的色谱峰,保留时间为26.578的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。(2)精密称取维生素c药物和维生素c适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中含维生素c0.1mg的溶液,作为供试品溶液;将维生素c稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;再分别将供试品溶液和对照品溶液注入高效液相色谱仪,记录色谱图,计算得出供试品溶液中维生素c的含量,完成维生素c的含量检测。(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。实施例3一种检测维生素c药物中维生素c及异维生素c含量的方法,所述高效液相色谱的检测条件为:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:30℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:0.8ml/min;检测器:紫外检测器;检测波长:268nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为80:20,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.1mol/l,缓冲盐溶液的ph值为6.8,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加入与实施例1相同稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c30μg的混合溶液,作为系统适用性试验溶液。精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图3和下表3:表3峰#保留时间峰高分离度理论塔板数对称因子异维生素c17.16856367——73281.03维生素c20.2161864553.86108171.09结合异维生素c与维生素c对照品的定位保留时间,图3看保留时间为17.168的色谱峰是异维生素c的色谱峰,保留时间为20.216的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。实施例4一种检测维生素c药物中维生素c及异维生素c含量的方法,所述高效液相色谱的检测条件为:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:30℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:1.2ml/min;检测器:紫外检测器;检测波长:268nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为80:20,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.1mol/l,缓冲盐溶液的ph值为7,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加入与实施例1相同稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c30μg的混合溶液,作为系统适用性试验溶液。精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图4和下表4:表4峰#保留时间峰高分离度理论塔板数对称因子异维生素c11.43854561——65111.04维生素c13.4681768923.5890041.06结合异维生素c与维生素c对照品的定位保留时间,图4看保留时间为11.438的色谱峰是异维生素c的色谱峰,保留时间为13.468的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。实施例5一种检测维生素c药物中维生素c及异维生素c含量的方法,所述高效液相色谱的检测条件为:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:30℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:1.0ml/min;检测器:紫外检测器;检测波长:268nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为80:20,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.05mol/l,缓冲盐溶液的ph值为6.8,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加入与实施例1相同稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c30μg的混合溶液,作为系统适用性试验溶液。精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图5和下表5:表5峰#保留时间峰高分离度理论塔板数对称因子异维生素c11.15350861——30511.39维生素c13.1741755702.7562991.07结合异维生素c与维生素c对照品的定位保留时间,图5看保留时间为11.153的色谱峰是异维生素c的色谱峰,保留时间为13.174的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。实施例6一种检测维生素c药物中维生素c及异维生素c含量的方法,所述高效液相色谱的检测条件为:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:25℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:1.0ml/min;检测器:紫外检测器;检测波长:210nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为80:20,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.1mol/l,缓冲盐溶液的ph值为5.5,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加入与实施例1相同稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c30μg的混合溶液,作为系统适用性试验溶液。精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图6和下表6:表6峰#保留时间峰高分离度理论塔板数对称因子异维生素c13.18148134——56950.98维生素c15.3871758873.3092501.07结合异维生素c与维生素c对照品的定位保留时间,图6看保留时间为13.181的色谱峰是异维生素c的色谱峰,保留时间为15.387的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。实施例7一种检测维生素c药物中维生素c及异维生素c含量的方法,所述高效液相色谱的检测条件为:高效液相色谱仪:沃特世e2695;色谱柱:hilic色谱柱,优选watersxbridgeamide,规格为4.6×250mm,5μm;柱温:40℃;流动相:缓冲盐溶液和乙腈的混合液,等度洗脱;流速:1.0ml/min;检测器:紫外检测器;检测波长:280nm;进样量:10μl。其中,所述混合液中缓冲盐溶液与乙腈的体积比为70:30,所述缓冲盐溶液为醋酸铵溶液,缓冲盐溶液的浓度为0.1mol/l,缓冲盐溶液的ph值为6.8,缓冲盐的ph值由氨水与醋酸或甲酸配合调节。所述的检测方法包括如下步骤:(1)分别精密称取维生素c药物与异维生素c的对照品适量,加入与实施例1相同稀释剂溶解制成每1ml中含维生素c0.1mg、异维生素c30μg的混合溶液,作为系统适用性试验溶液。精密量取系统适用性试验溶液10μl注入高效液相色谱仪,记录色谱图,异维生素c与维生素c色谱峰的分离度应不小于1.5,理论塔板数按维生素c峰计算不低于3000。具体结果见图7和下表7:表7峰#保留时间峰高分离度理论塔板数对称因子异维生素c6.493116018——5711——维生素c6.9883938041.574851.08结合异维生素c与维生素c对照品的定位保留时间,图7看保留时间为6.493的色谱峰是异维生素c的色谱峰,保留时间为6.988的色谱峰是维生素c的色谱峰。在上述色谱条件下,异维生素c与维生素c之间分离度好,且主峰峰形良好,分析时间短,满足中国药典的要求。(2)精密称取维生素c药物和维生素c对照品适量,分别加入步骤(1)所述稀释剂,将维生素c药物稀释成每1ml中分别含维生素c0.1mg、0.5mg的溶液,作为供试品溶液1#与供试品溶液2#;将维生素c对照品稀释成每1ml中含维生素c0.1mg的溶液作为对照品溶液;先分别精密量取供试品溶液1#和对照品溶液各10μl注入高效液相色谱仪,记录色谱图,按外标法以峰面积计算得出维生素c的含量;再精密量取供试品溶液2#10μl注入高效液相色谱仪,以面积归一化法计算得出异维生素c的含量。下面在实施例1的检测条件下分别对本方法的检测精密度和检测准确度进行试验,具体如下:1,检测精密度试验精密称取维生素c样品适量,加流动相溶解并制成每1ml中含维生素c0.1mg的溶液,作为测试溶液,将测试溶液注入高效液相色谱仪进行分析,进样6次,记录色谱图,结果见下表8:表8序号123456rsd%峰面积3554905353537935179503417426340613433615822.3%保留时间12.93212.89612.93412.88412.87512.8610.23%计算得到维生素c主峰峰面积的rsd为2.3%,保留时间的rsd为0.23%,证明本发明的检测方法精密度优异。2,检测准确度试验2.1维生素c含量准确度试验精密称取维生素c对照品适量,加流动相溶解并制成每1ml中含维生素c0.1mg,作为对照品溶液。精密称取维生素c对照品适量,加流动相溶解并制成每1ml中含维生素c1mg的溶液,作为准确度贮备溶液。然后按下表9分别准确移取贮备液0.8、1.0、1.2各1ml分别置10ml量瓶中,加流动相溶解并稀释至刻度,摇匀,即得准确度试验溶液,每个浓度水平平行配制3份准确度试验溶液。表9再分别取对照品溶液与准确度试验溶液注入高效液相色谱仪进行分析,记录色谱图,按外标法以峰面积计算测得量,结果见下表10:结论:维生素c含量准确度试验溶液r1~r3平均回收率为96.9%,符合要求(95.0%~105.0%),9个回收率数据的rsd值为0.64%,符合要求(rsd不得超过2%)。2.1异维生素c含量准确度试验精密称取异维生素c对照品适量,加流动相溶解并制成每1ml中含异维生素c0.5μg的溶液,作为对照品溶液,精密称取异维生素c对照品适量,加流动相溶解并制成每1ml中含异维生素c25μg的溶液,作为准确度贮备溶液。精密称取维生素c样品适量(含维生素c25mg),置50ml容量瓶中,然后按下表11分别准确移取贮备液0.8、1.0、1.2各1ml分别置50ml量瓶中,加流动相溶解并稀释至刻度,摇匀,即得准确度试验溶液,每个浓度水平平行配制3份准确度试验溶液。表11再分别取对照品溶液与准确度试验溶液注入高效液相色谱仪进行分析,记录色谱图,按外标法以峰面积计算测得量,结果见表12。表12异维生素c含量准确度结果结论:异维生素c含量准确度试验溶液r1~r3平均回收率为100.9%,符合要求(98.0%~102.0%),9个回收率数据的rsd值为1.2%,符合要求(rsd不得超过2%)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1