荧光增强型纳米探针及其制备方法以及岩石微纳米孔隙的光学显微镜成像方法与流程
本发明涉及地质资源和地质工程领域,具体而言,涉及一种荧光增强型纳米探针及其制备方法以及岩石微纳米孔隙的光学显微镜成像方法。
背景技术:
岩石孔隙结构表征是对地质资源进行精细化描述的基础,对正确认识岩石储层特征起指导性作用,同时,正确认识地质资源以及对其高效合理开发起决定作用。现有的运用复合纳米粒子表征岩石孔隙结构的材料主要是用包覆法合成,其实施过程是将纳米粒子和染料分子分散在正硅酸乙酯(teos)溶液中,然后加入无水乙醇使teos分解成一种类似网状结构的物质,在磁场条件下,将纳米探针注入到岩石孔隙之中,再在显微镜下通过观察探针的位置及其相互关系得到孔隙结构参数,利用探针占整个视域的百分比测算其孔隙度。
随着我国能源结构的不断调整,非常规油气(如页岩油气和致密油气)勘探开发逐步深入,其对应的岩石孔隙结构更为复杂,其岩石微纳米孔隙表征成为非常规油气研究的关键问题。然而,作为常规注入的用包覆方法合成的纳米探针存在发光强度低、稳定性差的问题。
技术实现要素:
本发明的目的在于提供一种荧光增强型纳米探针及其制备方法以及岩石微纳米孔隙的光学显微镜成像方法,以改善现有纳米探针存在的注入岩石后的发光强度低、稳定性差的问题。
本发明解决其技术问题是采用以下技术方案来实现的。
本发明提供了一种荧光增强型纳米探针,其包括:纳米粒子、二氧化硅改性层以及荧光染料,二氧化硅改性层附着于纳米粒子表面;荧光染料键合于二氧化硅改性层的表面。
本发明还提供了上述荧光增强型纳米探针的制备方法,其包括:将表面具有二氧化硅改性层的纳米粒子与荧光染料进行反应,以使得所述荧光染料键合于二氧化硅改性层。
本发明还提供了一种荧光增强型复合纳米粒子流体,其包括分散剂和上述荧光增强型纳米探针,可选地,分散剂为有机溶剂,有机溶剂包括乙醇、异丙醇、丙酮和乙二醇中至少一种,可选地,荧光增强型纳米探针在分散剂中的浓度为1×10-2~1×10-5mol/l。
本发明还提供了一种岩石微纳米孔隙的光学显微镜成像方法,其包括:将上述荧光增强型纳米探针注入岩石的裂缝和孔隙中;对充填有所述荧光增强型纳米探针的岩石的孔隙进行光学显微镜成像。
通过在纳米粒子的表面附着二氧化硅改性层,以使得其能够与荧光染料进行化学键合,进而荧光染料与纳米粒子以化学键合的方式结合在一起形成的荧光增强型纳米探针,其荧光性强,发光稳定。将该荧光增强型纳米探针注入岩石,荧光分子不易逸散,与岩石基质及胶结物颜色有明显差异,在光学显微镜下对于孔隙和喉道的识别非常有利,且该荧光增强型纳米探针能长期使用而不考虑荧光性随时间减弱的影响,可长期、准确、方便地反映出岩石的孔隙结构。
附图说明
为了更清楚地说明本发明实施方式的技术方案,下面将对实施方式中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
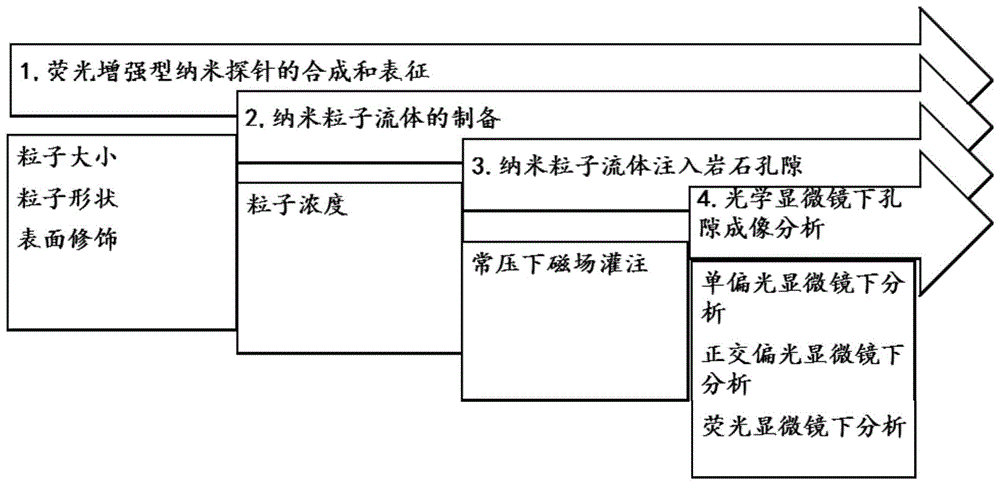
图1为本发明的实施方式的岩石微纳米孔隙的光学显微镜成像方法的流程图;
图2为本发明的实施例1的荧光增强型复合纳米粒子流体的扫描电镜图和场发射透射电子显微镜观察图;
图3为本发明的实施例2的荧光增强型复合纳米粒子流体的扫描电镜图和场发射透射电子显微镜观察图;
图4为本发明的实施例3的荧光增强型复合纳米粒子流体的扫描电镜图和场发射透射电子显微镜观察图;
图5为本发明的实施例4的键合法制备过程中不同的中间产物和荧光增强型纳米探针的对xrd衍射图进行分析后,通过origin软件处理后得到的数据分析图;
图6为本发明的实施例5的键合法制备过程中不同的中间产物和荧光增强型纳米探针采用振动样品磁强计(vsm)在室温下(300k)测量上述四种样品的磁滞回线;
图7为本发明的实施例6的荧光增强型纳米探针充填的岩石孔隙进行光学显微镜成像观察到的正交偏光和单片光下的照片;
图8为对比例1中的键合法与包覆法制备的纳米粒子的染料泄漏曲线;
图9为对比例2中的键合法与包覆法制备的纳米粒子充填的岩石孔隙进行光学显微镜成像在在暗场下(荧光光源)观察到的灌注键合法纳米粒子和灌注包覆法制备的纳米粒子的荧光照片。
具体实施方式
为使本发明实施方式的目的、技术方案和优点更加清楚,下面将对本发明实施方式中的技术方案进行清楚、完整地描述。实施方式或实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
下面对本发明实施方式的荧光增强型纳米探针及其制备方法以及岩石微纳米孔隙的光学显微镜成像方法进行具体说明。
本发明的一些实施方式提供了一种荧光增强型纳米探针,其包括:纳米粒子、二氧化硅改性层以及荧光染料,二氧化硅改性层附着于纳米粒子表面;荧光染料键合于二氧化硅改性层的表面。
现有技术中主要通过将纳米粒子和染料分子分散在正硅酸乙酯(teos)溶液中,然后加入无水乙醇使teos分解成一种类似网状结构的物质,在磁场条件下,将纳米探针注入到岩石孔隙之中,再在显微镜下通过观察探针的位置及其相互关系得到孔隙结构参数,利用探针占整个视域的百分比测算其孔隙度。然而,作为常规注入的用包覆方法合成的纳米探针存在发光强度低、稳定性差的问题。发明人经过研究发现造成其发光强度低、稳定性差的原因可能在于:包覆方法合成的纳米探针中,纳米粒子与染料分子以物理方式相结合,在高压作用注入岩石孔隙后,由于其粒径的原因使其染料分子结合数量有限;并且物理方式结合不牢固、染料分子容易逸散,从而造成稳定性差。因此,传统包覆法制备的复合纳米粒子无法满足长期使用的要求。
在此基础上发明人通过大量的实践和理论研究,以通过采用化学键合的方式来连接荧光染料,进而提高纳米粒子在岩石孔隙内的发光强度和稳定性。而进一步解决如何使得荧光染料与纳米粒子进行化学键合的问题,发明人通过在纳米粒子的表面附着能够与荧光染料进行化学键合的二氧化硅改性层,再将荧光染料键合在二氧化硅改性层的表面,进而达到化学结合荧光染料的目的。因此,由于荧光增强型磁性复合纳米粒子以键合方式与荧光染料分子结合,呈现黄绿色且荧光性更强,与岩石基质及胶结物颜色有明显差异,在光学显微镜下对于孔隙和喉道的识别非常有利,且荧光增强型磁性复合纳米粒子的荧光性更强,发光更稳定,能长期使用而不考虑荧光性随时间减弱的影响。进一步地,通过将荧光增强型纳米探针注入岩石的裂缝和孔隙中后,可以直接利用光学显微镜进行成像观察,即可长期、准确、方便地反映出岩石的孔隙结构。
根据一些实施方式,二氧化硅改性层的表面具有修饰得到的氨基,荧光染料通过酰胺反应于二氧化硅改性层键合。通过在二氧化硅的表面修饰氨基,进而使得其具有能够与具有羧基的荧光染料进行酰胺反应的基础。
进一步地,一些实施方式中,纳米粒子为磁性纳米粒子,可选地,磁性纳米粒子为fe3o4纳米粒子。磁性纳米粒子使得荧光增强型纳米探针能够具有磁性,进而能够在磁铁的作用下进行移动,便于通过磁铁将荧光增强型纳米探针简单方便的注入岩石孔隙中。
进一步地,一些实施方式中,荧光染料为罗丹明染料,可选地,罗丹明染料为罗丹明b。当然,其他实施方式中,荧光染料也可以是其他具有羧基或能够与氨基发生酰胺反应的其他荧光染料。
根据一些实施方式,荧光增强型纳米探针的粒径为15~180nm,例如荧光增强型纳米探针的粒径可以15~150nm,25~120nm或30~80nm等。在上述粒径范围内的纳米粒子既能够满足荧光增强型纳米探针能够很好地进入到岩石孔隙内的需求,又能够满足其在光学显微镜的观察下能够很好地进行成像与岩质进行区分。
本发明的一些实施方式还提供了一种上述荧光增强型纳米探针的制备方法,其包括:将表面具有二氧化硅改性层的所述纳米粒子与所述荧光染料进行反应,以使得荧光染料键合于所述二氧化硅改性层。
具体地,一些实施方式中,表面具有二氧化硅改性层的纳米粒子通过以下步骤制备得到:利用共沉淀法制备得到纳米粒子;在纳米粒子表面附着二氧化硅层,并对二氧化硅层进行表面修饰,形成二氧化硅改性层。一些实施方式中,对二氧化硅层进行修饰是通过表面化学修饰剂在二氧化硅层的表面修饰上氨基,其中,荧光染料为罗丹明b。
进一步地,一些实施方式中,表面化学修饰剂包括aptes(3-氨丙基三乙氧基硅烷)、aps(3-氨丙基乙氧基硅烷)、pei(聚醚酰亚胺)、ctab(十六烷基三甲基溴化铵)中的一种或两种以上的组合。例如,表面化学修饰剂可以为aptes(3-氨丙基三乙氧基硅烷),也可以为aps(3-氨丙基乙氧基硅烷),也可以为pei(聚醚酰亚胺),也可以为ctab(十六烷基三甲基溴化铵),或者表面化学修饰剂也可以为aptes(3-氨丙基三乙氧基硅烷)和aps(3-氨丙基乙氧基硅烷)的混合物,或pei(聚醚酰亚胺)和ctab(十六烷基三甲基溴化铵)的混合物等。当然,其他实施方式中,也可以选择其他能够对磁性二氧化硅复合粒子表面进行氨基修饰的表面修饰剂,不限于上述几种物质。
根据一些实施方式,纳米粒子为磁性纳米粒子,磁性纳米粒子的制备材料包括fecl2、feso4、fe(no3)2中的一种或几种,也可以选择例如fecl2、feso4的混合物,feso4、fe(no3)2的混合物等两种试剂的组合。当然,也可以是上述描述中三种或者三种以上的试剂的组合。同时,需要说明的是,在其他实施方式中,只要是能够满足用共沉淀法制备磁性纳米粒子的二价铁试剂均可以使用,并不限于上述列举的试剂。
根据一些实施方式,表面附着有二氧化硅层的纳米粒子即复合纳米粒子,二氧化硅层可以是包覆纳米粒子的二氧化硅壳层,用于与纳米粒子反应生成二氧化硅层的材料可为teos(正硅酸乙酯)。需要说明的是,在其他实施方式中,只要是能够满足在磁性纳米粒子表面包覆上sio2壳层的试剂均可以使用,并不限于上述列举的试剂。
进一步地,在共沉淀法制备得到纳米粒子后,添加表面活性剂对纳米粒子进行改性,表面活性剂包括檬酸三钠、壳聚糖、聚乙烯吡咯烷酮、聚对苯二甲酸乙二醇酯、硬脂酸、阿拉伯树胶、羟丙基甲基纤维素、海藻酸钠、十二烷基硫酸钠、十二烷基苯磺酸钠、聚乙烯醇、长链脂肪酸、淀粉和十二硫醇中的一种或两种以上的组合。通过上述表面活性剂能够避免形成的纳米粒子进行团聚,从而达到对荧光增强型纳米探针的粒径的控制。因此,当表面活性剂与纳米粒子的质量比降低时,合成的纳米粒子团聚较严重,并且形貌不均匀,提高表面活性剂与纳米粒子的质量比,可以减小荧光增强型纳米探针的粒径。
进一步地,一些实施方式中,将表面具有二氧化硅改性层的纳米粒子与荧光染料进行反应是在二氧化硅改性层表面键合荧光染料。具体地,在二氧化硅改性层表面键合荧光染料具体包括:将二氧化硅改性层和荧光染料在催化剂的作用下发生酰胺反应;进一步可选地,催化剂为nhs(n-羟基琥珀酰亚胺)和edc(1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐)。
一些实施方式,二氧化硅改性层和荧光染料进行酰胺反应是将所述荧光染料于表面具有二氧化硅改性层的纳米粒子的分散液中进行反应,从而使得其反应能够更加充分。分散纳米粒子的分散剂可以为无水乙醇,也可以为其他有机溶剂。根据一些实施方式,在荧光增强型纳米探针制备好后,对荧光增强型纳米探针的形貌和结构进行表征,优选荧光增强型纳米探针为球形或类球形,以使得其流动性更好。其形貌和结构的表征可以利用扫描电镜、透射电镜和x-射线衍射来完成。
本发明的一些实施方式还提供了一种荧光增强型复合纳米粒子流体,其包括分散剂和上述荧光增强型纳米探针。可选地,分散剂为有机溶剂,该有机溶剂包括乙醇、异丙醇、丙酮和乙二醇中至少一种。优选地,荧光增强型纳米探针在分散剂中的浓度为0.01~0.1g/ml,进而使得荧光增强型复合纳米粒子流体具有很好的流动性,能够比较容易地对岩石内部孔隙进行填充。
通过上述方法实现荧光增强型纳米探针的粒径控制、表面修饰、流体介质和粒子浓度等参数优化,以方便实现其纳米流体注入岩石之中。
根据一些实施方式,本发明还提供了一种岩石微纳米孔隙的光学显微镜成像方法,其包括:将上述荧光增强型纳米探针注入岩石的裂缝和孔隙中;对充填有所述荧光增强型纳米探针的岩石的孔隙进行光学显微镜成像。需要说明的是本发明的实施方式中的微纳米孔隙可以为15~1000nm的孔隙。
一些实施方式中,将荧光增强型纳米探针采用常压下利用磁场的方法注入到岩石的裂缝和孔隙中。
与常规表征岩石孔隙结构的铸体薄片法不同,高压灌注条件下和薄片的磨制会对岩石原始孔隙状态造成一定程度的破坏,且注入的试剂为大分子材料,即使在高压注入的条件下,仍然难以注入到某些微孔中去,制作过程复杂且周期较长;在本发明的实施方式中,将新型探针即荧光增强型探针填充到岩心孔隙中的方式为在常压下利用磁场将纳米粒子注入到岩石的裂缝和孔隙中。在常压下不会对岩石原始孔隙结构产生破坏;利用磁场进行填充也更为方便;纳米流体中纳米探针浓度的控制可以使纳米探针很大程度的填充进入岩石孔隙中的微孔隙以及微裂缝,最大程度的呈现出微纳米孔隙结构,且实施过程简单且一般实验室都能满足观察条件,避免了送样环节,缩短了实验周期。
具体地,在常压下利用磁场将纳米粒子对岩石孔隙进行填充的方法是,在常压下将荧光增强型纳米探针的流体至于岩石切片表面,将磁铁放置于岩石下方,再置于常压环境下,以使荧光增强型纳米探针随流体进入到岩石中。
根据一些实施方式,岩石的孔隙度为一般在2~30%,在将荧光增强型磁性复合纳米粒子注入岩石内之前,对岩石进行切片处理,优选地,将岩石岩心切片至厚度为30μm。
一些实施方式中,使用光学显微镜随机抽取岩石切片的30个以上的视域的孔隙和喉道进行分析。通过30个以上的视域能够充分科学地对岩石的孔隙结构进行反映,使得最后得到的结果更加准确。
参见附图1,本发明一些实施方式中提供的岩石微纳米孔隙的光学显微镜成像方法,先进行荧光增强型纳米探针的合成与表征,然后将荧光增强型纳米探针探分散在合适的流体中获得纳米粒子流体,然后在常压下利用磁场将探针注入岩石孔隙,再在光学显微镜下对荧光增强型纳米探针填充区域进行识别观察,借助单偏光与正交偏光进行所述岩石薄片鉴定,借助暗场下的荧光分析确定荧光增强型纳米探针填充区域,从而获得岩石的孔隙结构。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1
准确称取2.34gfecl3·6h2o(0.01mol)与0.96gfecl2·4h2o(0.005mol)于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)33ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物。将上述反应物分散到80ml的乙醇溶液中,加入25μlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。然后,取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。制备得到的纳米粒子。将制备得到的纳米粒子(平均粒径为36nm左右)分散于乙二醇中获得纳米粒子浓度为0.04g/ml的流体。
取适量的流体滴在导电胶上,待样品完全干燥后,固定于样品台,样品制好后表面喷金处理,通过场发射扫描电子显微镜(sem)观察,获得的照片如图2a;再取适量流体超声振荡10min,使样品尽量分散,形成比较均匀的悬浮液。将此悬浮液滴在碳膜网铜上,自然干燥后,通过场发射透射电子显微镜观察并获得的照片如图2b。
实施例2
准确称取2.00gfe2(so4)3(0.005mol)与0.76gfeso4(0.005mol)于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)55ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物。将上述反应物分散到80ml的乙醇溶液中,加入25μlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。然后,取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。制备得到的纳米粒子,如图2。将制备得到的纳米粒子(平均粒径为54nm左右)分散于乙醇中获得纳米粒子浓度为0.02g/ml的流体。
取适量的流体滴在导电胶上,待样品完全干燥后,固定于样品台,样品制好后表面喷金处理,通过场发射扫描电子显微镜(sem)观察,获得的照片如图3a;再取适量流体超声振荡10min,使样品尽量分散,形成比较均匀的悬浮液。将此悬浮液滴在碳膜网铜上,自然干燥后,通过场发射透射电子显微镜观察并获得的照片如图3b。
实施例3
准确称取2.34gfecl3·6h2o(0.01mol)与0.76gfeso4(0.005mol)于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)110ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物。将上述反应物分散到80ml的乙醇溶液中,加入25μlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。然后,取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。制备得到的纳米粒子,如图2。将制备得到的纳米粒子(平均粒径为62nm左右)分散于异丙醇中获得纳米粒子浓度为0.08g/ml的流体。
取适量的流体滴在导电胶上,待样品完全干燥后,固定于样品台,样品制好后表面喷金处理,通过场发射扫描电子显微镜(sem)观察,获得的照片如图4a;再取适量流体超声振荡10min,使样品尽量分散,形成比较均匀的悬浮液。将此悬浮液滴在碳膜网铜上,自然干燥后,通过场发射透射电子显微镜观察并获得的照片如图4b。
实施例4
a.fe3o4纳米粒子的制备
准确称取2.34gfecl3·6h2o(0.01mol)与0.96gfecl2·4h2o(0.005mol)于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。在真空干燥箱中干燥6h后,用玛瑙研钵将样品研细得到fe3o4纳米粒子粉末。
b.sio2纳米粒子的制备
采用改进的
c.fe3o4/sio2/aptes纳米粒子的制备
将上述反应物分散到80ml的乙醇溶液中,加入25μlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。得到fe3o4/sio2/aptes纳米粒子悬浮液。用磁铁分离后,放入真空干燥箱干燥后,用玛瑙研钵将样品研细得到fe3o4/sio2/aptes纳米粒子粉末。
d.fe3o4/sio2/aptes-rhb荧光增强型纳米探针的制备
将上述fe3o4/sio2/aptes纳米粒子粉末5.00mg加入到乙醇溶液分散后,超声处理1h,是样品在乙醇溶液中均匀分散。取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。干燥后研磨得到fe3o4/sio2/aptes-rhb荧光增强型纳米探针。
将上述制备的4种键合法制备过程中不同的中间产物(a.fe3o4b.sio2c.fe3o4/sio2/aptes纳米粒子)和最终产物d.fe3o4/sio2/aptes-rhb荧光增强型纳米探针,分别进行xrd(x射线衍射)表征,具体实施方式如下:
将样品粉末装填在样品板凹槽中,用一块光滑平整的玻璃片适当压实,将高出样品板平面的多余粉末刮去,重复几次,使样品表面平整。将样品板插入衍射仪样品台上,按操作程序启动仪器,对样品进行x射线扫描,得到衍射图。采用xrd分析软件jade5.0对xrd衍射图进行分析后,通过origin软件处理后得到的数据如图5所示,曲线a为制备的fe3o4磁性纳米粒子的xrd图谱,衍射峰位置和相对强度与pdf#88-0315标准卡片fe3o4基本相符,出现了明显的(220)、(311)、(400)、(422)、(511)、(440)等特征衍射峰,表明制备出的磁性纳米粒子物相为反尖晶石结构,峰型较尖锐,纳米粒子结晶较完整。曲线b为采用改进的
实施例5
a.fe3o4纳米粒子的制备
准确称取2.34gfecl3·6h2o(0.01mol)与0.96gfecl2·4h2o(0.005mol)于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。在真空干燥箱中干燥6h后,用玛瑙研钵将样品研细得到fe3o4纳米粒子粉末。
b.fe3o4/sio2纳米粒子的制备
将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)110ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物fe3o4/sio2纳米粒子。干燥后,用玛瑙研钵将样品研细得到fe3o4/sio2纳米粒子粉末。
c.fe3o4/sio2/aptes纳米粒子的制备
将上述反应物分散到80ml的乙醇溶液中,加入25μlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。得到fe3o4/sio2/aptes纳米粒子悬浮液。用磁铁分离后,放入真空干燥箱干燥后,用玛瑙研钵将样品研细得到fe3o4/sio2/aptes纳米粒子粉末。
d.fe3o4/sio2/aptes-rhb荧光增强型磁性复合纳米粒子的制备
将上述fe3o4/sio2/aptes纳米粒子粉末5.00mg加入到乙醇溶液分散后,超声处理1h,是样品在乙醇溶液中均匀分散。取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。干燥后研磨得到fe3o4/sio2/aptes-rhb荧光增强型纳米探针。
对上述制备的4种键合法制备过程中不同的中间产物(a.fe3o4b.fe3o4/sio2,c.fe3o4/sio2/aptes纳米粒子)和最终产物d.fe3o4/sio2/aptes-rhb荧光增强型纳米探针,分别进行磁性能测试,具体实施方式如下:
采用振动样品磁强计(vsm)在室温下(300k)测量上述四种样品的磁滞回线,得到的数据如图6所示。从图中可以看出,制备的fe3o4(曲线a)磁性粒子的最大饱和磁化强度(ms)为54.2emu/g,经sio2包覆及后续的氨基修饰和rhb染料的键合后,fe3o4/sio2(曲线b),fe3o4/sio2/aptes(曲线c),fe3o4/sio2/aptes-rhb(曲线d)纳米粒子的磁化强度有所降低。但粒子的矫顽力和剩磁都趋于零,说明所制备的纳米粒子均有较好的超顺磁性。
实施例6
准确称取2.34gfecl3·6h2o与0.96gfecl2·4h2o于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)110ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物。将上述反应物分散到80ml的乙醇溶液中,加入25mlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。然后,取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。将制备得到的纳米粒子(平均粒径为62nm左右)分散于乙二醇中获得纳米粒子浓度为0.04g/ml的流体。
待分析岩心先进行岩石切片处理,制备岩石薄片,先将所述岩石岩心置于薄片切割机(petro-thin)上切割并磨成130μm的厚度,再把130μm的岩石岩心薄片利用真空原理固定在真空吸盘上,然后直接在薄片研磨盘(lappingplate)上研磨至标准厚度30μm。然后,将荧光增强型纳米探针置于岩石薄片上,将磁铁放置于岩石下方,再置于常压环境下,以使荧光增强型纳米探针随流体进入到岩石薄片中。用酒精润湿的棉球将表面多余的纳米粒子清理干净后,对纳米粒子充填的岩石孔隙进行光学显微镜成像。观察到的正交偏光(图7a)和单片光下(图7b)的照片如图7所示。通过岩石切片的正交照片和单片光的照片,可以很好的分析岩石的矿物颗粒的分布和种类的判别,对岩石进行分类。
对比例1
键合法制备fe3o4/sio2/aptes-rhb荧光增强型纳米探针:
准确称取2.34gfecl3·6h2o与0.96gfecl2·4h2o于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)110ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物。将上述反应物分散到80ml的乙醇溶液中,加入25mlaptes搅拌反应3h。用磁铁将合成的fe3o4/sio2/aptes纳米粒子从反应体系中分离,用无水乙醇洗涤3次,调节溶液ph为7.0。然后,取5ml浓度为5×10-4mol/lrhb、0.3834gedc和0.23018gnhs加入到80mlfe3o4/sio2/aptes乙醇溶液中,室温下搅拌反应5h。用磁铁将磁性荧光纳米材料与溶液分离,先用乙醇洗涤3次后,再用去离子水洗涤,到肉眼观察上清液为无色为止。将制备得到的纳米粒子(平均粒径为62nm左右)分散于乙二醇中获得纳米粒子浓度为0.04g/ml的流体。
包覆法制备fe3o4/sio2/rhb磁性荧光纳米粒子
准确称取2.34gfecl3·6h2o与0.96gfecl2·4h2o于三颈烧瓶中,加100ml去离子水(通氮气除氧)搅拌溶解,放入恒温水浴锅后将温度加热到80℃,在氮气的保护和机械搅拌下,向混合溶液中缓慢滴加40ml1.15mol/l的naoh溶液,待滴加完毕后,加入40ml0.1mol/l柠檬酸三钠作为表面活性剂,恒温80℃搅拌0.5h后,用磁倾析法将合成的fe3o4纳米粒子从反应体系中分离。将制备的fe3o4纳米粒子重新分散在80%乙醇溶液中,加入5mlnh3·h2o,5ml浓度为5×10-4mol/lrhb后,在氮气的保护和机械搅拌下,用恒压滴液漏斗向反应中滴加teos乙醇溶液(vteos/v无水乙醇=1/10)110ml,继续搅拌反应2h。用乙醇多次清洗后,得到反应产物fe3o4/sio2/rhb磁性荧光纳米粒子。将制备得到的纳米粒子(平均粒径为68nm左右)分散于乙二醇中获得纳米粒子浓度为0.04g/ml的流体。
图8为键合法与包覆法制备的纳米粒子的染料泄漏曲线,我们对比了包覆法制备的68nm和键合法制备的62nm的粒子在保存过程中的染料泄漏情况,随着时间的推移,包覆法制备的纳米粒子的荧光强度的损失较大,这是由于物理包覆的染料分子在保存或使用过程中容易从介孔二氧化硅壳层中逃逸出来。在相同条件下,通过共价键结合染料的键合法制备的纳米粒子由于具有更强的结合力,染料分子不易丢失,在保存或使用过程中能保持有比较稳定的荧光强度。同时从图中可以看出在粒径分布相似的情况下,键合法制备的复合粒子有更强的荧光。
对比例2
取对比例1中键合法制备的62nm左右的荧光增强型磁性纳米粒子和包覆法制备的68nm左右的磁性荧光纳米粒子的流体,将所述荧光增强型磁性复合纳米粒子置于对比例1所述岩石薄片上,将磁铁放置于所述岩石下方,再置于常压环境下,以使所述键合法和包覆法制备的纳米粒子随流体进入到所述岩石薄片中。用酒精润湿的棉球将表面多余的纳米粒子清理干净后,对纳米粒子充填的岩石孔隙进行光学显微镜成像。在暗场下(荧光光源)观察到的灌注键合法纳米粒子和灌注包覆法制备的纳米粒子的荧光照片如图9所示。可以看到在相同的仪器条件下(放大倍数、曝光时间等),键合法纳米粒子的荧光更明亮,在光学显微镜荧光下呈现黄绿色,成像效果更好可以很方便的观察到岩石孔隙的存在。而包覆法的纳米粒子的荧光却较微弱,不能很好地分辨和观察岩石的孔隙结构。样品的主要孔隙大多为粒间孔,几乎没有粒内孔存在。通过图像分析软件imagej对获得的随机选取30个区域进行观察的照片进行分析,获得岩石平均面孔率为12.36%。
综上所述,由于荧光增强型纳米探针在光学显微镜下产生黄绿色荧光,与岩石基质的岩石差异非常明显,对于孔隙和喉道的识别非常有利,且荧光增强型磁性复合纳米粒子荧光性更强、发光更稳定因而能够保存更长时间;纳米粒子能够通过控制teos的用量调节粒子的粒径大小,获得形貌形状大小相似、粒径分布较窄的粒子,在光学显微镜下更加明显地区别于天然岩石基质;纳米粒子能够进行可控的表面修饰,控制表面的带电状态和极性等,有助于改善纳米流体和岩石的界面状况,方便纳米粒子注入岩石孔隙之中;本发明实施方式中只需纳米粒子、岩石薄片样品的制作,再利用光学显微镜观察即可。实施过程简单且一般实验室都能满足观察条件,避免了送样环节,缩短了实验周期。
以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!