一种启脾丸的鉴别方法与流程
本发明涉及中医药检测技术领域,具体是一种启脾丸的鉴别方法。
背景技术:
启脾丸由人参、麸炒白术、茯苓、甘草、陈皮、山药、莲子(炒)、炒山楂、六神曲(炒)、炒麦芽、泽泻等十一味组成,十一味药均以细粉混以炼蜜制成丸剂直接入药。在《中国药典》2010第二增补本、2015年版一部和内蒙古天奇国家局标准ybz15682009(水丸)等三个现行标准均收载了启脾丸的显微鉴别和薄层鉴别。但现行的标准中,药典标准中只对茯苓、人参、山药、陈皮、甘草、炒山楂六味药材进行显微质量控制,还有药典已有显微标准的麸炒白术、莲子(炒)、炒麦芽和泽泻共四种药材未进行控制。而且薄层鉴别也存在供试品斑点不清晰,供试品与对照品rf值不一致等缺点。
收录于《中国药典》2010年版第二增补本白术的鉴别为:取样品9g,加硅藻土5g,研细,加乙醚30ml,加热回流30分钟,滤过,滤液挥干,残渣加正己烷2ml使溶解,作为供试品溶液,展开剂为石油醚(60~90℃)-乙酸乙酯(50:1)。在实验过程中发现该方法提取的启脾丸样品,经显色后苍术酮斑点不清晰且rf值较大。
《中国药典》2015年版一部现行陈皮的鉴别方法为:取样品6g,剪碎,加硅藻土4g,研匀,加甲醇加热回流1小时,滤过,滤液作为供试品溶液。在实验过程中发现,由于启脾丸样品中含有大量的蜂蜜,直接将滤液作为供试品溶液在实验过程中极易出现拖尾或rf值与对照品不一致的现象。
泽泻是泽泻科植物多年生水生草本东方泽泻的干燥块茎,泽泻具有独特的药用及保健功能,大量用于保健品及中成药的生产中,药材市场经常出现不少混伪品。经试验,用薄层色谱图法检测启脾丸模拟样品未发现23-乙酰泽泻醇b对应斑点;按中国药典2015版一部“泽泻”【含量测定】项下方法进行实验,启脾丸模拟样品中未发现与23-乙酰泽泻醇b对照品相对应的峰;采用特征图谱方法,启脾丸处方中甘草和陈皮等药材杂质峰干扰大,难以有效分离,因此暂无法建立泽泻的hplc鉴别方法;即现有的技术无法准确检测出启脾丸中的泽泻成分。六神曲具有健脾和胃、消食调中的作用,用于脾胃虚弱、饮食停滞、胸痞腹胀、小儿食积。各省炮制规范或中药材标准均有收载,其处方组成均包含面粉、麦麸、苦杏仁、赤小豆、青蒿、辣蓼、苍耳草,但各药材剂量配比不尽相同,出入较大。启脾丸处方中六神曲投料仅为所有药材投入量的十分之一,而苦杏仁在六神曲中所占比例也很小。由于苦杏仁苷在样品中含量低,使用一般的常量鉴别方法难以鉴别出来,采用薄层色谱法进行鉴别,薄层色谱中未发现启脾丸与苦杏仁苷对照品相应位置显相同斑点;因此,需要使用灵敏度较高的方法进行鉴别。
技术实现要素:
本发明的目的是提供一种启脾丸的鉴别方法,修订了山楂的显微特征,增加了白术、炒麦芽和泽泻的显微特征,完善了启脾丸成分的显微特征;对白术和陈皮的薄层色谱法进行了改进,解决了现有方法中,白术经显色后斑点不清晰且rf值较大的问题以及陈皮极易出现拖尾或rf值与对照品不一致的现象;采用高效液相色谱-质谱法鉴别泽泻、六神曲成分,灵敏度和准确性较高。
为了实现上述目的,所述一种启脾丸的鉴别方法包括以下内容:
(1)在显微镜下观察启脾丸的表面性状,观察结果如下:
1)茯苓:不规则分枝状团块无色,遇水合氯醛试液溶化;菌丝无色或淡棕色,直径4~6µm。2)人参:草酸钙簇晶直径20~68µm,棱角锐尖。
3)山药:草酸钙针晶束存在于黏液细胞中,长80~240µm,针晶直径2~8µm。
4)陈皮:草酸钙方晶成片存在于薄壁组织中。
5)甘草:纤维束周围薄壁细胞含草酸钙方晶,形成晶纤维。
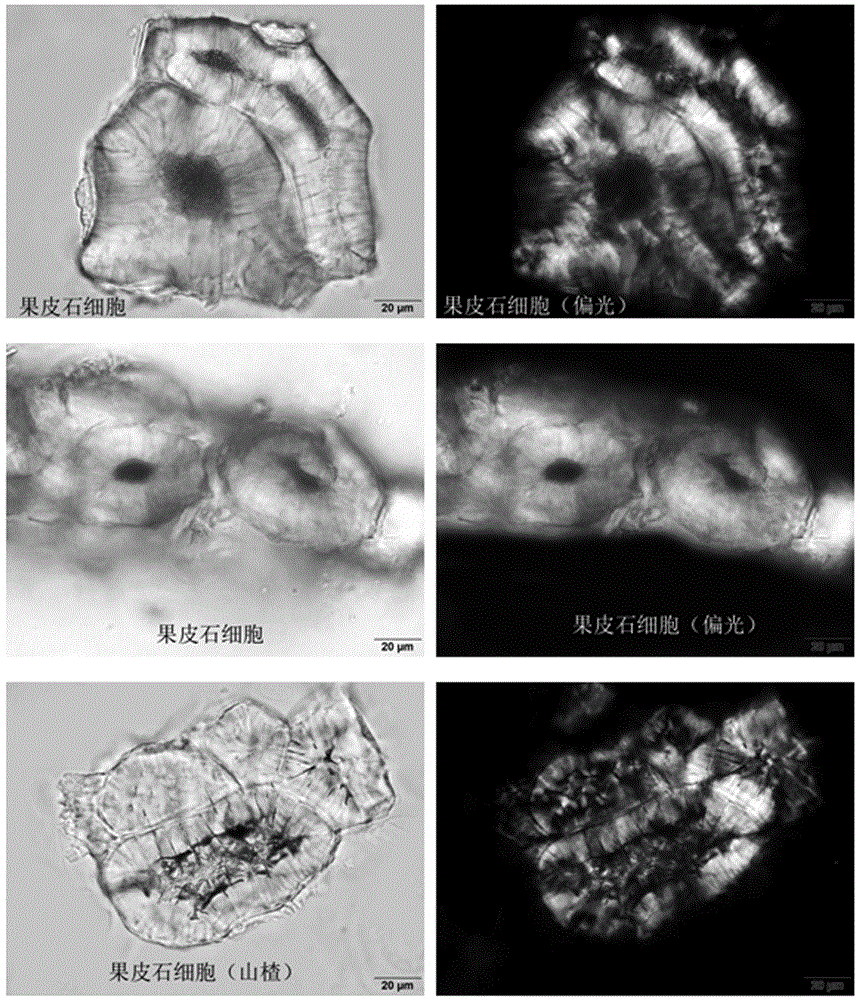
6)山楂:果皮石细胞单个散在或成群,无色或淡黄色,类多角形、长圆形或不规则形,直径19~125μm,壁厚,孔沟及层纹明显,有的胞腔内含深棕色物。
7)白术:草酸钙针晶细小,长10~32μm,存在于薄壁细胞中。
8)炒麦芽:稃片外表皮表面观长细胞与2个短细胞(栓化细胞、硅质细胞)交互排列;长细胞壁厚,紧密深波状弯曲,短细胞类圆形,有稀疏壁孔。
9)泽泻:薄壁细胞类圆形,具多数椭圆形纹孔,集成纹孔群。
(2)用薄层色谱法鉴别白术成分,具体内容如下:
1)取启脾丸样品9g,置挥发油提取器中,加水100ml连接挥发油提取器,自测定器上端加水至充满测定器的刻度部分并溢流入烧瓶时为止,再加入乙酸乙酯3ml,加热回流3小时,放冷,分离取乙酸乙酯层,作为供试品溶液。
2)取白术对照药材0.5g,置挥发油提取器中,加水100ml连接挥发油提取器,自测定器上端加水至充满测定器的刻度部分并溢流入烧瓶时为止,再加入乙酸乙酯3ml,加热回流3小时,放冷,分离取乙酸乙酯层,即得对照药材溶液。
3)吸取供试品溶液、对照药材溶液各10μl,分别点于同一硅胶g板上,以石油醚(60~90℃)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸显色剂,置日光下检视。供试品色谱中,在与对照药材色谱相应的位置上,显有一桃红色主斑点(苍术酮)。
(3)用薄层色谱法鉴别陈皮成分,具体内容如下:
1)取启脾丸样品6g,剪碎,加70%乙醇20ml,加热回流1小时,滤过,滤液加在聚酰胺柱(30~60目,5g,柱内径为1.5cm,干法装柱)上,先用80ml水洗脱,弃去水洗液,再用70%乙醇100ml洗脱,收集乙醇洗脱液,回收溶剂至干,残渣加甲醇10ml使溶解,作为供试品溶液。
2)取陈皮对照药材1g,剪碎,加70%乙醇20ml,加热回流1小时,滤过,滤液加在聚酰胺柱(30~60目,5g,柱内径为1.5cm,干法装柱)上,先用80ml水洗脱,弃去水洗液,再用70%乙醇100ml洗脱,收集乙醇洗脱液,回收溶剂至干,残渣加甲醇10ml使溶解,得到对照药材溶液。
3)取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。
4)吸取供试品溶液、对照药材溶液、对照品溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水(32:17:5)的下层溶液为展开剂,展开,取出,晾干,喷以三氯化铝试液,置紫外灯(365nm)下检视。供试品色谱中,在与对照药材及对照品色谱相应的位置上,显相同颜色的荧光斑点。
所述白术成分和陈皮成分的薄层色谱鉴别均还包括进行耐用性实验考察和制备阴性样品溶液进行方法学验证。
(4)采用高效液相色谱-质谱法鉴别泽泻成分,具体内容如下:
1)色谱条件:以十八烷基硅烷键合硅胶为填充剂(色谱柱内径为2.1mm),以0.1%甲酸为流动相a,甲醇为流动相b,按表1中的规定进行梯度洗脱;流速为每分钟0.5ml。
2)质谱条件:采用质谱检测器,电喷雾正离子模式(esi+),进行多反应检测(mrm),选择质荷比m/z555.4→495.3作为24-乙酰泽泻醇a检测离子对,选择质荷比m/z537.4→491.2和质荷比m/z537.4→105.1作为23-乙酰泽泻醇b检测离子对,选择质荷比551.4→477.3和质荷比m/z551.4→57.1作为23-乙酰泽泻醇c检测离子对。取对照品溶液,进样2μl,上述检测离子对的mrm色谱峰的信噪比均应大于3:1。
3)供试品溶液制备:取启脾丸样品剪碎,混匀,取2g,加硅藻土1g,研匀,加甲醇50ml,超声处理30分钟,滤过,滤液浓缩至5ml,滤过,取续滤液,即得。
4)对照品溶液制备:取24-乙酰泽泻醇a、23-乙酰泽泻醇b和23-乙酰泽泻醇c对照品,加甲醇制成每1ml含各对照品各1μg的混合标准溶液,作为对照品溶液。
5)测定:吸取对照品溶液和供试品溶液各2μl,注入高效液相色谱-串联质谱联用仪,测定。
6)结果判定:以质荷比m/z555.4→495.3离子对提取的供试品离子流色谱中,应出现与24-乙酰泽泻醇a对照品色谱保留时间一致的色谱峰;以质荷比m/z537.4→491.2和质荷比m/z537.4→105.1离子对提取的供试品离子流色谱中,应出现与23-乙酰泽泻醇b对照品色谱保留时间一致的色谱峰;以质荷比m/z551.4→477.3和质荷比m/z551.4→57.1离子对提取的供试品离子流色谱中,应出现与23-乙酰泽泻醇c对照品色谱保留时间一致的色谱峰。
(5)采用高效液相色谱-质谱法鉴别六神曲成分,具体内容如下:
1)色谱条件:以十八烷基硅烷键合硅胶为填充剂(色谱柱内径为2.1mm),以0.1%甲酸为流动相a,乙腈为流动相b,按表2中的规定进行梯度洗脱;流速为每分钟0.5ml。
2)质谱条件:采用质谱检测器,电喷雾正离子模式(esi+),进行多反应检测(mrm),选择质荷比m/z475.2→325.2、质荷比m/z475.2→163.1和质荷比m/z475.2→145.0作为苦杏仁苷检测离子对。取对照品溶液,进样2μl,上述检测离子对的mrm色谱峰的信噪比均应大于3:1。
3)取苦杏仁苷对照品,加甲醇制成浓度为0.1μg/ml的溶液,作为对照品溶液。
4)取样品剪碎,混匀,取2g,加硅藻土1g,研匀,加甲醇50ml,超声处理30分钟,滤过,滤液浓缩至5ml,滤过,取续滤液,即得供试品溶液。
5)吸取对照品溶液和供试品溶液各2μl,注入高效液相色谱-串联质谱联用仪,测定。
6)结果判定:以质荷比m/z475.2→325.2、质荷比m/z475.2→163.1和质荷比m/z475.2→145.0离子对提取的供试品离子流色谱中,应出现与苦杏仁苷对照品色谱保留时间一致的色谱峰。
以上所述泽泻成分、六神曲成分的高效液相色谱-质谱法也适用于启脾口服液中泽泻成分、六神曲成分的鉴别,其中启脾口服液供试品溶液的制备方法为:将启脾口服液过0.22μm滤膜,即得。其他步骤与启脾丸的鉴别方法一致。
本发明的有益效果:
本发明通过在显微镜下观察启脾丸的表面性状,修订了山楂的显微特征,增加了白术、炒麦芽和泽泻的显微特征,完善了启脾丸成分的显微特征。
本发明改进了白术成分薄层色谱鉴别中供试品溶液的制备方法,将启脾丸置挥发油提取器中,加水连接挥发油提取器,自测定器上端加水至充满测定器的刻度部分并溢流入烧瓶时为止,再加入乙酸乙酯,加热回流3小时,放冷,分离取乙酸乙酯层,得到鉴别所需供试品溶液。通过改进供试品溶液的提取方法,解决了常规方法提取的样品溶液经显色后苍术酮斑点不清晰且rf值较大的问题。
本发明中用薄层色谱法鉴别陈皮成分的内容参考了《中国药典》2015年版一部启脾口服液橙皮苷的鉴别方法,通过聚酰胺柱将样品溶液中的蜂蜜去除,解决了由于启脾丸样品中含有大量的蜂蜜,直接将滤液作为供试品溶液在实验过程中极易出现拖尾或rf值与对照品不一致的现象;使得样品中橙皮苷与对照品的rf值一致,同时将启脾丸与启脾口服液中橙皮苷鉴别的方法统一。在本发明的方法中同时加入陈皮对照药材作为对照药材溶液与供试品溶液进行比较,增加鉴别方法的可靠性。白术成分和陈皮成分的薄层色谱鉴别中还进行耐用性实验考察和制备阴性样品溶液进行方法学验证,耐用性好、鉴别结果准确。
本发明通过高效液相色谱-质谱法鉴别泽泻成分,解决了泽泻薄层色谱鉴别方法、hplc鉴别方法中存在23-乙酰泽泻醇b难以检出、干扰大等问题,可以准确地检测出泽泻成分中24-乙酰泽泻醇a、23-乙酰泽泻醇b和23-乙酰泽泻醇c的含量,对启脾丸和启脾口服液中泽泻的质量控制具有重要意义。
本发明通过灵敏度较高的高效液相色谱-质谱法鉴别六神曲成分,解决了由于苦杏仁苷在启脾丸样品中含量低,使用一般的常量鉴别方法难以鉴别出来的问题,对提高六神曲的质量、控制启脾丸和启脾口服液中六神曲的投料具有重要意义。
本发明通过显微镜鉴别启脾丸中所有药材的成分,再通过薄层色谱法鉴别启脾丸中白术和陈皮的成分,最后利用高效液相色谱-质谱法鉴别泽泻和六神曲成分,通过多种方法完善了现有技术中启脾丸的鉴别标准,为控制启脾丸的投料提供了技术基础。
附图说明
图1是启脾丸样品山楂药材果皮石细胞的显微镜图片;
图2是启脾丸样品白术药材石细胞的显微镜图片;
图3是启脾丸样品薄壁细胞中白术草酸钙针晶的显微镜图片;
图4是启脾丸样品炒麦芽药材稃片长细胞与2个短细胞的显微镜图片;
图5是启脾丸样品泽泻药材薄壁细胞的显微镜图片;
图6是10个厂家启脾丸样品考察的白术薄层色谱图,图中1表示启脾丸(h公司,批号:1604),2表示启脾丸(e公司,批号:a16280),3表示启脾丸(b制药厂,批号:4500050),4表示启脾丸(i制药厂,批号:1605118),5表示启脾丸(d公司,批号:1711042),6表示启脾丸(g公司,批号:11180201),7表示启脾丸(g公司,批号:12180401),8表示启脾丸(c公司,批号:170801),9表示启脾丸(j制药厂,批号:16013870),10表示启脾丸(f公司,批号:160105),11表示启脾丸(a公司,批号:818058),12表示启脾丸模拟样,13表示白术对照药材(中检院,批号:120925-201611),14表示苍术酮(stanfordchemicals,批号:pf180502-04),a表示桃红色斑点;各样品的点样量是10μl,苍术酮的点样量是1μl;
图7是启脾丸样品分别经药典原方法与本发明方法提取供试品溶液并鉴别白术成分得到的薄层色谱对比图,图中1表示启脾丸(j制药厂,批号:15013034)用药典原方法提取(点样量为30μl),2表示启脾丸(j制药厂,批号:15013034)用本发明方法提取(点样量为5μl),3表示白术对照药材(点样量为5μl),a表示桃红色斑点;
图8是使用青岛海洋化工有限公司提供的硅胶g板,批号20160715,在温度30℃、湿度70%rh条件下检视得到的白术薄层色谱图,图中1表示缺白术阴性样品,2表示白术对照药材(中检院,批号:120925-201611),3表示启脾丸(j制药厂,批号:16013870),4表示启脾丸(g公司,批号:12180401),5表示启脾丸(g公司,批号:11180201),a表示桃红色斑点;点样量均为10μl;
图9是使用烟台市化学工业研究所提供的硅胶g板,批号20170413,在温度30℃、湿度70%rh条件下检视得到的白术薄层色谱图,图中1表示缺白术阴性样品,2表示白术对照药材(中检院,批号:120925-201611),3表示启脾丸(j制药厂,批号:16013870),4表示启脾丸(g公司,批号:12180401),5表示启脾丸(g公司,批号:11180201),a表示桃红色斑点;点样量均为10μl;
图10是使用merck公司提供的硅胶g板,批号hx85448326,在温度30℃、湿度70%rh条件下检视得到的白术薄层色谱图,图中1表示缺白术阴性样品,2表示白术对照药材(中检院,批号:120925-201611),3表示启脾丸(j制药厂,批号:16013870),4表示启脾丸(g公司,批号:12180401),5表示启脾丸(k公司,批号:11180201),a表示桃红色斑点;点样量均为10μl;
图11是使用青岛海洋化工有限公司提供的硅胶g板,批号20160715,在温度4℃、湿度60%rh条件下检视得到的白术薄层色谱图,图中1表示缺白术阴性样品,2表示白术对照药材(中检院,批号:120925-201611),3表示启脾丸(j制药厂,批号:16013870),4表示启脾丸(g公司,批号:12180401),5表示启脾丸(g公司,批号:11180201),a表示桃红色斑点;点样量均为10μl;
图12是使用青岛海洋化工有限公司提供的硅胶g板,批号20160715,在温度28℃、湿度32%rh条件下检视得到的白术薄层色谱图,图中1表示缺白术阴性样品,2表示白术对照药材(中检院,批号:120925-201611),3表示启脾丸(j制药厂,批号:16013870),4表示启脾丸(g公司,批号:12180401),5表示启脾丸(g公司,批号:11180201),a表示桃红色斑点;点样量均为10μl;
图13是对白术薄层鉴别点样量考察的色谱图,图中1~4启脾丸(j制药厂,批号:16013870)的点样量分别为2、5、10、15μl;5~8白术对照药材的点样量分别为2、5、10、15μl;a表示桃红色斑点;
图14是10个厂家启脾丸样品考察的陈皮薄层色谱图,图中1表示启脾丸(c公司,批号:171001),2表示启脾丸(b制药厂,批号:4500044),3表示启脾丸(j制药厂,批号:16013870),4表示启脾丸(e公司,批号:a15282),5表示启脾丸(f公司,批号:160105),6表示启脾丸(a公司,批号:816043),7表示启脾丸(g公司,批号:11161122),8表示启脾丸(d公司,批号:1610021),9表示启脾丸(h公司,批号:1605),10表示启脾丸(g公司,批号:12170909),11表示启脾丸(i制药厂,批号:1703166),12表示橙皮苷;13表示启脾丸模拟样,14表示陈皮对照药材,a表示黄绿色荧光斑点;点样量均为10μl;
图15是启脾丸模拟样用药典原方法与本发明的方法提取供试品溶液并对陈皮成分进行鉴别得到的薄层色谱对比图,图中1表示模拟样按药典原标准方法提取,2表示陈皮对照药材按药典原标准方法提取,3表示模拟样按本发明方法提取,4表示陈皮对照药材按本发明方法提取,5表示橙皮苷,a表示黄绿色荧光斑点;点样量均为10μl;
图16是使用青岛海洋化工有限公司提供的硅胶g板,批号20160715,在温度30℃、湿度75%rh条件下检视得到的陈皮薄层色谱图,图中1表示缺陈皮阴性样品,2表示橙皮苷,3表示启脾丸(c公司,批号:171001),4表示启脾丸(b制药厂,批号:4500044),5表示启脾丸(j制药厂,批号:16013870),6表示陈皮对照药材,a表示黄绿色荧光斑点;点样量均为10μl;
图17是使用烟台市化学工业研究所提供的硅胶g板,批号20170413,在温度30℃、湿度75%rh条件下检视得到的陈皮薄层色谱图,图中1表示缺陈皮阴性样品,2表示橙皮苷,3表示启脾丸(c公司,批号:171001),4表示启脾丸(b制药厂,批号:4500044),5表示启脾丸(j制药厂,批号:16013870),6表示陈皮对照药材,a表示黄绿色荧光斑点;进样量均为10μl;
图18是使用merck公司提供的硅胶g板,批号hx85448326,在温度30℃、湿度75%rh条件下检视得到的陈皮薄层色谱图,图中1表示缺陈皮阴性样品,2表示橙皮苷,3表示启脾丸(c公司,批号:171001),4表示启脾丸(b制药厂,批号:4500044),5表示启脾丸(j制药厂,批号:16013870),6表示陈皮对照药材,a表示黄绿色荧光斑点;点样量均为10μl;
图19是使用青岛海洋化工有限公司提供的硅胶g板,批号20160715,在温度4℃、湿度40%rh条件下检视得到的陈皮薄层色谱图,图中1表示缺陈皮阴性样品,2表示橙皮苷,3表示启脾丸(c公司,批号:171001),4表示启脾丸(b制药厂,批号:4500044),5表示启脾丸(j制药厂,批号:16013870),6表示陈皮对照药材,a表示黄绿色荧光斑点;点样量均为10μl;
图20是使用青岛海洋化工有限公司提供的硅胶g板,批号20160715,在温度30℃、湿度32%rh条件下检视得到的陈皮薄层色谱图,图中1表示缺陈皮阴性样品,2表示橙皮苷,3表示启脾丸(c公司,批号:171001),4表示启脾丸(b制药厂,批号:4500044),5表示启脾丸(j制药厂,批号:16013870),6表示陈皮对照药材,a表示黄绿色荧光斑点;点样量均为10μl;
图21是陈皮成分鉴别点样量考察的薄层色谱图,图中1~4橙皮苷的点样量分别为2、5、10、15μl,5~8陈皮对照药材的点样量分别为2、5、10、15μl,9~12启脾丸(c公司,批号:171001)的点样量分别为2、5、10、15μl;a表示黄绿色荧光斑点;
图22是24-乙酰泽泻醇a、23-乙酰泽泻醇b和23-乙酰泽泻醇c混合对照品的hplc-msmrm图;
图23是启脾丸样品(f公司,批号:160105)的hplc-msmrm图;
图24是启脾口服液样品(k公司,批号:20171155)的hplc-msmrm图;
图25是缺泽泻阴性样品的hplc-msmrm图;
图26是苦杏仁苷对照品的hplc-msmrm图;
图27是启脾丸样品(d公司,批号:1711042)的hplc-msmrm图;
图28是启脾口服液样品(k公司,批号:20171155)的hplc-msmrm图;
图29是缺苦杏仁阴性样品的hplc-msmrm图。
具体实施方式
为了更加详细的介绍本发明,下面结合实施例,对本发明做进一步说明。
一种启脾丸的鉴别方法包括以下内容:
(1)在显微镜下观察启脾丸的表面性状,观察结果如下:
1)茯苓:不规则分枝状团块无色,遇水合氯醛试液溶化;菌丝无色或淡棕色,直径4~6µm。2)人参:草酸钙簇晶直径20~68µm,棱角锐尖。
3)山药:草酸钙针晶束存在于黏液细胞中,长80~240µm,针晶直径2~8µm。
4)陈皮:草酸钙方晶成片存在于薄壁组织中。
5)甘草:纤维束周围薄壁细胞含草酸钙方晶,形成晶纤维。
6)山楂:果皮石细胞单个散在或成群,无色或淡黄色,类多角形、长圆形或不规则形,直径19~125μm,壁厚,孔沟及层纹明显,有的胞腔内含深棕色物,见图1。同时因为处方中白术药材也有石细胞显微特征,但是显微特征有区别“石细胞淡黄色,类圆形、多角形、长方形或少数纺锤形,直径37~64μm。”白术石细胞壁较薄,胞腔较大,胞腔内无内容物,见图2。
7)白术:草酸钙针晶细小,长10~32μm,存在于薄壁细胞中,见图3。白术草酸钙针晶细小,散在薄壁细胞中,与山药的草酸钙针晶束差异特征大。
8)炒麦芽:稃片外表皮表面观长细胞与2个短细胞(栓化细胞、硅质细胞)交互排列;长细胞壁厚,紧密深波状弯曲,短细胞类圆形,有稀疏壁孔,见图4。
9)泽泻:薄壁细胞类圆形,具多数椭圆形纹孔,集成纹孔群,见图5。在启脾丸检品中均可见到距集成纹孔群的薄壁细胞,波状弯曲的内皮层细胞少见;所以只收录泽泻的薄壁细胞特征。
(2)用薄层色谱法鉴别白术成分,具体内容如下:
1)取启脾丸样品9g,置挥发油提取器中,加水100ml连接挥发油提取器,自测定器上端加水至充满测定器的刻度部分并溢流入烧瓶时为止,再加入乙酸乙酯3ml,加热回流3小时,放冷,分离取乙酸乙酯层,作为供试品溶液。
2)取白术对照药材0.5g,置挥发油提取器中,加水100ml连接挥发油提取器,自测定器上端加水至充满测定器的刻度部分并溢流入烧瓶时为止,再加入乙酸乙酯3ml,加热回流3小时,放冷,分离取乙酸乙酯层,即得对照药材溶液。
3)吸取供试品溶液、对照药材溶液各10μl,分别点于同一硅胶g板上,以石油醚(60~90℃)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸显色剂,置日光下检视。供试品色谱中,在与对照药材色谱相应的位置上,显有一桃红色主斑点(苍术酮)。采用本发明的方法对10个厂家的启脾丸样品进行了试验,结果10个厂家样品均检出白术,10个厂家启脾丸样品考察的白术薄层色谱图见图6。将本发明所述方法与药典原方法进行对比见图7。
4)耐用性实验考察:①用不同厂家生产的薄层硅胶g板进行考察,见图8~10;②对不同展开温度:4℃、30℃进行考察,见图8、图11;③对不同相对湿度32%、70%进行考察,见图8、图12。④对对照药材溶液和启脾丸样品(j制药厂,批号:16013870)溶液的点样量进行考察,分别点样2、5、10、15μl,结果显示当点样量为10μl时,对照药材色谱与样品色谱均能得到清晰的斑点,分离效果良好,见图13。结果表明本薄层色谱耐用性良好。
5)方法学验证:按处方比例称取除白术外其余药材,并按启脾丸制法制成的缺白术阴性样品,取缺白术阴性样品适量,按步骤1)供试品溶液制备方法制成阴性样品溶液,按步骤3)进行点样,展开,显色,结果阴性样品色谱中,在与对照药材色谱相应的位置上,未显相同颜色的斑点,见图8~12,表明该法阴性无干扰,符合方法学要求。
(3)用薄层色谱法鉴别陈皮成分,具体内容如下:
1)取启脾丸样品6g,剪碎,加70%乙醇20ml,加热回流1小时,滤过,滤液加在聚酰胺柱(30~60目,5g,柱内径为1.5cm,干法装柱)上,先用80ml水洗脱,弃去水洗液,再用70%乙醇100ml洗脱,收集乙醇洗脱液,回收溶剂至干,残渣加甲醇10ml使溶解,作为供试品溶液。
2)取陈皮对照药材1g,剪碎,加70%乙醇20ml,加热回流1小时,滤过,滤液加在聚酰胺柱(30~60目,5g,柱内径为1.5cm,干法装柱)上,先用80ml水洗脱,弃去水洗液,再用70%乙醇100ml洗脱,收集乙醇洗脱液,回收溶剂至干,残渣加甲醇10ml使溶解,得到对照药材溶液。
3)取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。
4)吸取供试品溶液、对照药材溶液、对照品溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水(32:17:5)的下层溶液为展开剂,展开,取出,晾干,喷以三氯化铝试液,置紫外灯(365nm)下检视。供试品色谱中,在与对照药材及对照品色谱相应的位置上,显相同颜色的荧光斑点。采用本发明的方法对10个厂家启脾丸样品进行试验,结果10个厂家样品均检出陈皮,10个厂家启脾丸样品考察的陈皮薄层色谱图见图14,将本发明所述方法与药典原方法进行对比见图15。
5)耐用性实验考察:①用不同厂家生产的薄层硅胶g板进行考察,见16~18图;②对不同展开温度:4℃、30℃进行考察,见图16、图19;③对不同相对湿度32%、75%进行考察,见图16、图20;④对对照品溶液、对照药材溶液和启脾丸样品(c公司,批号:171001)溶液的点样量进行考察,分别点样2、5、10、15μl,结果显示当点样量为10μl时,对照药材色谱与样品色谱均能得到清晰的斑点,分离效果良好,见图21。结果表明本薄层色谱耐用性良好。
6)方法学验证:取缺陈皮的阴性样品适量,按步骤1)供试品溶液制备方法制成阴性样品溶液,按步骤4)点样,展开,显色,结果阴性样品色谱中,在与对照药材色谱相应的位置上,未显相同颜色的斑点,见图16~20,表明该法阴性无干扰,符合方法学要求。
(4)采用高效液相色谱-质谱法鉴别泽泻成分,具体内容如下:
1)色谱条件:以十八烷基硅烷键合硅胶为填充剂(色谱柱内径为2.1mm),以0.1%甲酸为流动相a,甲醇为流动相b,按表1中的规定进行梯度洗脱;流速为每分钟0.5ml。
2)质谱条件:质谱装配电喷雾电离源(esi),干燥气温度:200℃;雾化器压力:20psi;干燥气流速:12l/min,鞘流气温度:350℃;鞘流气流速:11l/min,highpressurerf:150v;lowpressurerf:60v;毛细管电压:4000v;喷嘴电压:500v;驻留时间:60ms;检测模式:正离子检测,扫描方式:多反应监测(mrm)。取对照品溶液,进样2μl,上述检测离子对的mrm色谱峰的信噪比均应大于3:1。质谱参数见表3。
3)供试品溶液制备:取启脾丸样品剪碎,混匀,取2g,加硅藻土1g,研匀,加甲醇50ml,超声处理30分钟,滤过,滤液浓缩至5ml,滤过,取续滤液,即得。
4)对照品溶液制备:取24-乙酰泽泻醇a、23-乙酰泽泻醇b和23-乙酰泽泻醇c对照品,加甲醇制成每1ml含各对照品各1μg的混合标准溶液,作为对照品溶液。
5)测定:吸取对照品溶液和供试品溶液各2μl,注入高效液相色谱-串联质谱联用仪,测定。
6)结果判定:以质荷比m/z555.4→495.3离子对提取的供试品离子流色谱中,应出现与24-乙酰泽泻醇a对照品色谱保留时间一致的色谱峰;以质荷比m/z537.4→491.2和质荷比m/z537.4→105.1离子对提取的供试品离子流色谱中,应出现与23-乙酰泽泻醇b对照品色谱保留时间一致的色谱峰;以质荷比m/z551.4→477.3和质荷比m/z551.4→57.1离子对提取的供试品离子流色谱中,应出现与23-乙酰泽泻醇c对照品色谱保留时间一致的色谱峰。结果如图22~23所示。
以上所述泽泻成分的高效液相色谱-质谱法也适用于启脾口服液中泽泻成分的鉴别,其中启脾口服液供试品溶液的制备方法为:将启脾口服液过0.22μm滤膜,即得。其他步骤与启脾丸的鉴别方法一致。结果见图24。经检测,10批来自不同生产单位的启脾丸和1批启脾口服液均检出24-乙酰泽泻醇a、23-乙酰泽泻醇b和23-乙酰泽泻醇c有成分。
所述方法还进行了方法学验证:取缺泽泻的阴性样品适量,按步骤3)供试品溶液制备方法制成阴性样品溶液,按色谱-质谱条件进样进行检测,结果阴性样品的hplc-msmrm图中,未发现与对照品相对应的峰,见图25,表明该法阴性无干扰,符合方法学要求。
(5)采用高效液相色谱-质谱法鉴别六神曲成分,具体内容如下:
1)色谱条件:以十八烷基硅烷键合硅胶为填充剂(色谱柱内径为2.1mm),以0.1%甲酸为流动相a,乙腈为流动相b,按表2中的规定进行梯度洗脱;流速为每分钟0.5ml。
2)质谱条件:采用质谱检测器,电喷雾正离子模式(esi+),进行多反应检测(mrm),选择质荷比m/z475.2→325.2、质荷比m/z475.2→163.1和质荷比m/z475.2→145.0作为苦杏仁苷检测离子对。取对照品溶液,进样2μl,上述检测离子对的mrm色谱峰的信噪比均应大于3:1。
3)取苦杏仁苷对照品,加甲醇制成浓度为0.1μg/ml的溶液,作为对照品溶液。
4)取样品剪碎,混匀,取2g,加硅藻土1g,研匀,加甲醇50ml,超声处理30分钟,滤过,滤液浓缩至5ml,滤过,取续滤液,即得供试品溶液。
5)吸取对照品溶液和供试品溶液各2μl,注入高效液相色谱-串联质谱联用仪,测定。
6)结果判定:以质荷比m/z475.2→325.2、质荷比m/z475.2→163.1和质荷比m/z475.2→145.0离子对提取的供试品离子流色谱中,应出现与苦杏仁苷对照品色谱保留时间一致的色谱峰。结果见图26~27。
以上所述六神曲成分的高效液相色谱-质谱法也适用于启脾口服液中六神曲成分的鉴别,其中启脾口服液供试品溶液的制备方法为:将启脾口服液过0.22μm滤膜,即得。其他步骤与启脾丸的鉴别方法一致。结果见图28。经检测,10批来自不同生产单位的启脾丸和1批启脾口服液均检出有苦杏仁苷成分。
所述方法还进行了方法学验证:取缺苦杏仁苷的阴性样品适量,按步骤4)供试品溶液制备方法制成阴性样品溶液,按色谱-质谱条件进样进行检测,结果阴性样品的hplc-msmrm图中,未发现与对照品相对应的峰,见图29,表明该法阴性无干扰,符合方法学要求。
以上所述泽泻和六神曲成分检测中所检测的样品来自11家生产单位,样品信息见表4。
- 还没有人留言评论。精彩留言会获得点赞!