新铃兰醛的制备方法及其在化妆品检测中的用途与流程
本发明属于化妆品技术领域,涉及一种化妆品的检测方法,尤其涉及一种检测化妆品尤其是化妆品中致敏性芳香剂的检测方法,以及该种芳香剂的制备方法,所制得的芳香剂可以方便地应用于化妆品中该种芳香剂的检测或者用作某些产品的添加剂。特别地,所述致敏性芳香剂是新铃兰醛。
背景技术:
据统计,化妆品过敏中至少有35%是由香料引起的过敏。针对可能由于使用香料所引发的问题,各主要相关国际机构与组织出台了相应的法规政策以对香料香精市场进行约束。香精香料直接或间接地与“人体健康”相关联,成为各国政府强制管理的对象。欧盟化妆品法规(ecno.1223/2009-eucosmeticsregulation-inforce)根据医院相关过敏数据的统计结果规定:符合“在不可冲洗型化妆品中含量大于等于0.001%;在可冲洗型化妆品中含量大于等于0.01%”条件时,化妆品香料中26种致敏原物质必须在标签上标注(在欧盟发布的化妆品指令2005年修订前规定香料成分免于标签标注,仅以“香料或香精”标出即可),借此来提示对此类物质过敏人群购买时应谨慎。
国家质检总局发布通告,欧盟委员会2017年8月2日发布(eu)2017/1410号法规,修订化妆品法规(ec)no1223/2009的附录ii禁用物质清单和附录iii限用物质清单。其中禁用物质清单中增加了新铃兰醛(两个同分异构体)、苔黑醛、氯化苔黑醛3种化学物质,同时删除附录iii化妆品限用物质清单第79条款关于新铃兰醛(hicc)的内容。(eu)2017/1410号修订案内容将于2017年8月23日起生效(其中,删除附录iii化妆品限用物质清单第79条款自2021年8月23日起生效)。这3种物质在香水中被广泛使用,包括原始版本香奈儿5号、迪奥的迪奥小姐等,但曾引起多起过敏。
新铃兰醛是一种香气不同寻常、价值极高的含氧萜烯类合成香料,它具有持久的铃兰花香香韵和清淡而甜润的兔耳草花香气,并有丰富的余香,香气温和而稳定,其柔和的花香似羟基香茅醛,但强度更大。由于新铃兰醛在香精配方中的作用显著,越来越受到重视,已经在香水香精等各种香精中逐渐取代了传统的铃兰花香型合成香料铃兰醛的位置。
尽管香料过敏以及禁用的法规仅在欧盟地区执行,但我国已对香料致敏原引起关注,并进行了致敏原斑贴试验以及定性定量检测研究等工作,且很多化妆品商品成分标识上也明确标注了致敏原。新增加的禁用香料国内尤其要引起重视。目前国内外关于香精香料的分析检测有一些报道,常见的检测方法主要有气相色谱法、气-质联用法、液-质联用法等,涉及的香精成分也主要是致敏原香料,样品前处理方式主要为振荡溶剂萃取、超声辅助溶剂提取等。而对于化妆品中新铃兰醛残留量的气相色谱-三重四极杆质谱法测定的方法研究,国内外文献及标准中几乎没有报道。多级质谱与单级质谱相比,定性更准确,抗干扰能力强,可从干扰严重的一级碎片中抽取母离子,进一步碎裂得到子离子来定量,降低了样品基质背景的响应,灵敏度也高于单级质谱,更适合于复杂基质中微量、痕量物质的检测。
新铃兰醛,英文名称为lyral,为对(间)-(4-羟基-4-甲基戊基)-3-环己烯-1-甲醛同分异构体的混合物,分子式为c12h22o2,分子量为210.32,化学结构式为式(i)和式(ii),亦可分别称为新铃兰醛i和新铃兰醛ii或者新铃兰醛1和新铃兰醛2。新铃兰醛是一种重要的精细化学品,为无色粘性液体,具有清淡而甜润的兔耳草花香,有较好的铃兰香韵,香气稳定而持久。新铃兰醛在日化香精中应用广泛,可用于配制高级香水、香皂、洗涤剂、化妆品等日化产品的香精。
新铃兰醛的合成路线主要有两种途径:其一:月桂烯水合生成月桂烯醇,水合时先使月桂烯与二氧化硫发生反应生成环状的月桂烯砜,以保护月桂烯分子结构中的共轭双键;然后在酸催化作用下使月桂烯砜进行水合反应,再加热脱去二氧化硫,得到中间体月桂烯醇;月桂烯醇再与丙烯醛进行diels-alder反应。据目前现有报道看,该方案的月桂烯醇收率(以月桂烯计)最高只有70%。其二:月桂烯先与丙烯醛进行diels-alder反应生成柑青醛,柑青醛再水合反应生成新铃兰醛。该方案中月桂烯醇收率(以月桂烯计)最高只有32.56%。还有些其他合成工艺的报道,但收率更低,没有工业化意义。
上述第一种途径中,月桂烯砜脱二氧化硫过程时,月桂烯醇易脱水和聚合,导致月桂烯醇收率难超过70%,因此极大影响了新铃兰醛的收率。上述第二种途径中,月桂烯醇与丙烯醛进行diels-alder反应时,通常需要加催化剂,投料量达3~5%(以月桂烯醇计)原料成本增加了,而且需要将催化剂先溶解好再投入,增加操作的复杂性;3)催化剂不能回收利用,变成了危险的废弃物,增加处理费用。
美国专利文献us4007137公开了一种热压法制备新铃兰醛的方法,主要包括以下步骤:将月桂烯醇、丙烯醛和对苯二酚加入到密闭高压釜中,在150℃下反应4.5h,得到新铃兰醛。由于月桂烯醇和丙烯醛为剧烈放热反应,会使反应体系急剧升温导致丙烯醛挥发,因此反应需要在高压、密闭条件下进行,不仅对反应设备要求较高,而且增加了新铃兰醛的成本。该文献还公开了一种路易斯酸催化法制备新铃兰醛的方法,主要包括以下步骤:将月桂烯醇、路易斯酸催化剂和有机溶剂混合后升温,然后加入丙烯醛,反应1h~10h后得到新铃兰醛,其中,路易斯酸催化剂一般为氯化锌、溴化锌、四氯化锡等,有机溶剂一般为甲苯。路易斯酸催化法虽然能够降低反应温度,但是需要对反应产物进行后处理,如去除催化剂、去除有机溶剂等,不仅操作步骤繁琐,而且会产生大量废酸腐蚀设备、污染环境。
cn109734568a(201910059012.4)公开了一种新铃兰醛的制备方法,其步骤依次为将月桂烯醇砜与碱和阻聚剂按照重量比1000︰3~5︰1~3混配,在120~125℃、-0.03~-0.09mpa下预热1~10秒;在125~135℃、-0.03~-0.09mpa下裂解1~10秒,得含月桂烯醇的裂解物;在釜温135~150℃、釜压-0.03~-0.09mpa、顶温70~100℃、顶压-0.03~-0.09mp条件下精馏处理,收集含有月桂烯醇和月桂烯的塔顶出料;与丙烯醛在不添加催化剂下加成反应,再减压精馏,得新铃兰醛。据信该发明采用月桂烯醇砜裂解,提高月桂烯醇的单程转化收率,从而提高总收率,降低副产物。
cn102531858b(201010592887.x)公开了一种新铃兰醛的制备方法,包括以下步骤:a)将月桂烯醇升温至50℃~130℃;b)向步骤a)得到的月桂烯醇中滴加丙烯醛,滴加过程中控制温度为50℃~130℃,滴加完毕后继续反应,得到新铃兰醛。据信该发明通过控制丙烯醛的加入方式和加入温度,使月桂烯醇和丙烯醛无需在高温高压或者在路易斯酸催化剂和有机溶剂的条件下发生diels-alder反应,反应条件温和,副产物较少。由于无需高温高压,因此对反应设备要求较低,不会增加新铃兰醛的成本;由于无需加入路易斯酸催化剂,省却了繁琐的后处理工序,也不会产生大量废酸腐蚀设备、污染环境。
在-化妆品检测中,通常需要提供新铃兰醛对照品。一方面,这种对照品价格昂贵、不易获得,例如通常需要从国外购买;另一方面,由于新铃兰醛是挥发性油状物,作为对照品使用不方便,例如在精密取样时非常不方便,也容易造成不准确的问题;再一方面,因新铃兰醛的挥发性问题而造成对照品在长期贮藏或者多次使用过程中容易因挥发而造成对照品取样不准确,进而造成-化妆品检测不准确的问题。
因此,提供一种适合作为检测用新铃兰醛对照品仍是本领域技术人员迫切期待的,提供制备这种新铃兰醛对照品的方法亦是本领域技术人员迫切期待的。
技术实现要素:
本发明的目的在于提供一种适合作为检测用的新铃兰醛对照品,本发明另一目的在于提供制备这种新铃兰醛对照品的方法,本发明的再一目的在于提供一种以新铃兰醛为功能成分的香料添加剂,已经出人意料的发现,采用本发明方法获得的新铃兰醛对照品,呈现优异的理化特征,并且尤其适用作为-化妆品检测用的对照品;此外,本发明所得添加了氯化钾的新铃兰醛,鉴于其稳定性尤其是挥发稳定性大大提高,并且其中的氯化钾是一种化学惰性且生理学常用的物质,这种物料同样使得新铃兰醛广泛适用于其它领域,例如在可以添加氯化钾的情况下作为香料添加剂。本发明基于此类发现而得以完成。
为此,本发明第一方面提供了制备新铃兰醛对照品的方法,其包括如下步骤:
(a)在对苯二酚存在下,向控制温度在110℃~130℃范围内的月桂烯醇中缓缓滴加丙烯醛,接着使反应充分完成;
(b)将所得反应混合物用去离子水洗涤,得到新铃兰醛粗品,对该粗品进行气相色谱分析以确定其中新铃兰醛的含量大于85%;
(c)将所得新铃兰醛粗品在真空度2mmhg以下进行精馏,收集120℃~125℃之间的新铃兰醛馏分,对该新铃兰醛馏分进行气相色谱分析以确定其中新铃兰醛的含量大于99.0%,为精制的新铃兰醛;
(d)使所得新铃兰醛馏分导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:180~220,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此混合物置于-25~-18℃的温度下冷冻8~12小时,再使其回复到室温,通过摇晃使粘连的细颗粒物分散,接着将此分散的细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量,即得可用作对照品检测用的新铃兰醛。
根据本发明第一方面的方法,其中步骤(a)中,对苯二酚用量以重量计是月桂烯醇添加量的0.08~0.12%,例如0.1%。
根据本发明第一方面的方法,其中步骤(a)中,丙烯醛用量以摩尔计是月桂烯醇添加量的1~1.2倍。
根据本发明第一方面的方法,其中步骤(a)中,每3.5~4.2mol丙烯醛滴加的时间为5~7小时,例如约6小时。
根据本发明第一方面的方法,其中步骤(a)中,丙烯醛滴加完毕后,继续进行反应0.5~2小时例如约1小时以使反应充分完成。
根据本发明第一方面的方法,其中步骤(a)是照如下方式进行的:向装有温度计、机械搅拌装置和回流冷凝器的1000ml四口烧瓶中加入539g(3.5mol)月桂烯醇和0.539g对苯二酚,加热至110℃;向所述月桂烯醇中滴加3.85mol~4.2mol丙烯醛,滴加6h,滴加过程中,将所述反应瓶置于冷冻槽中,保持反应瓶温度在110℃~130℃;丙烯醛滴加完毕后,继续反应1h,得到反应混合物。
根据本发明第一方面的方法,其中步骤(b)中,每100g物料使用23~26g去离子水洗涤,洗涤2~4次,例如3次。
根据本发明第一方面的方法,其中步骤(d)中,所述氯化钾的纯度大于99.5%。例如所用氯化钾符合2015年版《中国药典》二部第1374页的要求。
根据本发明第一方面的方法,其中步骤(d)中,新铃兰醛与氯化钾的重量比为100:200。
根据本发明第一方面的方法,其中步骤(d)中,采用气相色谱分析标定细颗粒物中新铃兰醛的含量,是采用已知含量的新铃兰醛标准品进行标定,该已知含量的新铃兰醛通常是市售可得的。
根据本发明第一方面的方法,测定各种物料中新铃兰醛含量所用气相色谱法是气相色谱-三重四级杆串联质谱测定法。
根据本发明第一方面的方法,其中步骤(d)中,经冷冻处理所得分散的细颗粒物的流动性以休止角表示,其休止角为48°~52°。在本发明中,如未另外说明,参数“休止角”的测定方法参见教科书《药剂学》(奚念朱主编,第三版,人民卫生出版社出版,1996年4月第3版,isbn7-117-00026-0)第248-249页描述的方法,具体使用第249页第3-5行所述的“固定漏斗法”。需要说明的是,表征或者测定本发明组合物颗粒/粉末休止角的方法有许多,例如在奚念朱主编的第三版《药剂学》教科书还列举了其它一些测定方法。在本发明上下文中,如果未另外说明,表征或者测定本发明组合物颗粒/粉末休止角的方法是使用上述“固定漏斗法”进行的。已经出人意料地发现,通过本发明,将油状新铃兰醛与大约2倍量的氯化钾细颗粒混合,可以得到具有一定流动性的细颗粒物,这为新铃兰醛作为标准品应用提供了极佳的物理性能,例如有利于称量和样品处理。另外,由于氯化钾的惰性,以及后续作为对照品测定时的样品处理过程中,氯化钾极易除去进而不会影响测定结果的准确性。另外,如本文所详细记载的,本发明所得呈细颗粒物形式的新铃兰醛,比之于油状新铃兰醛具有明显更好的稳定性,例如在敞口环境中的新铃兰醛颗粒物的挥发性远远小于油状新铃兰醛。
进一步的,本发明第二方面的提供了一种新铃兰醛对照品,其为新铃兰醛与氯化钾以重量比100:180~220比例的混合物。
根据本发明第二方面的新铃兰醛对照品,其为呈分散的细颗粒物形式。
根据本发明第二方面的新铃兰醛对照品,其为呈分散的细颗粒物形式,该细颗粒物的流动性以休止角表示,其休止角为48°~52°。
根据本发明第二方面的新铃兰醛对照品,其是照包括如下步骤的方法制备得到的:
(a)在对苯二酚存在下,向控制温度在110℃~130℃范围内的月桂烯醇中缓缓滴加丙烯醛,接着使反应充分完成;
(b)将所得反应混合物用去离子水洗涤,得到新铃兰醛粗品,对该粗品进行气相色谱分析以确定其中新铃兰醛的含量大于85%;
(c)将所得新铃兰醛粗品在真空度2mmhg以下进行精馏,收集120℃~125℃之间的新铃兰醛馏分,对该新铃兰醛馏分进行气相色谱分析以确定其中新铃兰醛的含量大于99.0%,为精制的新铃兰醛;
(d)使所得新铃兰醛馏分导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:180~220,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此混合物置于-25~-18℃的温度下冷冻8~12小时,再使其回复到室温,通过摇晃使粘连的细颗粒物分散,接着将此分散的细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量,即得可用作对照品检测用的新铃兰醛。
根据本发明第二方面的新铃兰醛对照品,其中步骤(a)中,对苯二酚用量以重量计是月桂烯醇添加量的0.08~0.12%,例如0.1%。
根据本发明第二方面的新铃兰醛对照品,其中步骤(a)中,丙烯醛用量以摩尔计是月桂烯醇添加量的1~1.2倍。
根据本发明第二方面的新铃兰醛对照品,其中步骤(a)中,每3.5~4.2mol丙烯醛滴加的时间为5~7小时,例如约6小时。
根据本发明第二方面的新铃兰醛对照品,其中步骤(a)中,丙烯醛滴加完毕后,继续进行反应0.5~2小时例如约1小时以使反应充分完成。
根据本发明第二方面的新铃兰醛对照品,其中步骤(a)是照如下方式进行的:向装有温度计、机械搅拌装置和回流冷凝器的1000ml四口烧瓶中加入539g(3.5mol)月桂烯醇和0.539g对苯二酚,加热至110℃;向所述月桂烯醇中滴加3.85mol~4.2mol丙烯醛,滴加6h,滴加过程中,将所述反应瓶置于冷冻槽中,保持反应瓶温度在110℃~130℃;丙烯醛滴加完毕后,继续反应1h,得到反应混合物。
根据本发明第二方面的新铃兰醛对照品,其中步骤(b)中,每100g物料使用23~26g去离子水洗涤,洗涤2~4次,例如3次。
根据本发明第二方面的新铃兰醛对照品,其中步骤(d)中,所述氯化钾的纯度大于99.5%。例如所用氯化钾符合2015年版《中国药典》二部第1374页的要求。
根据本发明第二方面的新铃兰醛对照品,其中步骤(d)中,新铃兰醛与氯化钾的重量比为100:200。
根据本发明第二方面的新铃兰醛对照品,其中步骤(d)中,采用气相色谱分析标定细颗粒物中新铃兰醛的含量,是采用已知含量的新铃兰醛标准品进行标定,该已知含量的新铃兰醛通常是市售可得的。
根据本发明第二方面的新铃兰醛对照品,测定各种物料中新铃兰醛含量所用气相色谱法是气相色谱-三重四级杆串联质谱测定法。
进一步的,本发明第三方面的提供了一种组合物,其为新铃兰醛与氯化钾以重量比100:180~220比例的混合物。
根据本发明第三方面的组合物,其为呈分散的细颗粒物形式。
根据本发明第三方面的组合物,其为呈分散的细颗粒物形式,该细颗粒物的流动性以休止角表示,其休止角为48°~52°。
根据本发明第三方面的组合物,其是照包括如下步骤的方法制备得到的:
(a)在对苯二酚存在下,向控制温度在110℃~130℃范围内的月桂烯醇中缓缓滴加丙烯醛,接着使反应充分完成;
(b)将所得反应混合物用去离子水洗涤,得到新铃兰醛粗品,对该粗品进行气相色谱分析以确定其中新铃兰醛的含量大于85%;
(c)将所得新铃兰醛粗品在真空度2mmhg以下进行精馏,收集120℃~125℃之间的新铃兰醛馏分,对该新铃兰醛馏分进行气相色谱分析以确定其中新铃兰醛的含量大于99.0%,为精制的新铃兰醛;
(d)使所得新铃兰醛馏分导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:180~220,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此混合物置于-25~-18℃的温度下冷冻8~12小时,再使其回复到室温,通过摇晃使粘连的细颗粒物分散,接着将此分散的细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量,即得。
根据本发明第三方面的组合物,其中步骤(a)中,对苯二酚用量以重量计是月桂烯醇添加量的0.08~0.12%,例如0.1%。
根据本发明第三方面的组合物,其中步骤(a)中,丙烯醛用量以摩尔计是月桂烯醇添加量的1~1.2倍。
根据本发明第三方面的组合物,其中步骤(a)中,每3.5~4.2mol丙烯醛滴加的时间为5~7小时,例如约6小时。
根据本发明第三方面的组合物,其中步骤(a)中,丙烯醛滴加完毕后,继续进行反应0.5~2小时例如约1小时以使反应充分完成。
根据本发明第三方面的组合物,其中步骤(a)是照如下方式进行的:向装有温度计、机械搅拌装置和回流冷凝器的1000ml四口烧瓶中加入539g(3.5mol)月桂烯醇和0.539g对苯二酚,加热至110℃;向所述月桂烯醇中滴加3.85mol~4.2mol丙烯醛,滴加6h,滴加过程中,将所述反应瓶置于冷冻槽中,保持反应瓶温度在110℃~130℃;丙烯醛滴加完毕后,继续反应1h,得到反应混合物。
根据本发明第三方面的组合物,其中步骤(b)中,每100g物料使用23~26g去离子水洗涤,洗涤2~4次,例如3次。
根据本发明第三方面的组合物,其中步骤(d)中,所述氯化钾的纯度大于99.5%。例如所用氯化钾符合2015年版《中国药典》二部第1374页的要求。
根据本发明第三方面的组合物,其中步骤(d)中,新铃兰醛与氯化钾的重量比为100:200。
根据本发明第三方面的组合物,其中步骤(d)中,采用气相色谱分析标定细颗粒物中新铃兰醛的含量,是采用已知含量的新铃兰醛标准品进行标定,该已知含量的新铃兰醛通常是市售可得的。
根据本发明第三方面的组合物,测定各种物料中新铃兰醛含量所用气相色谱法是气相色谱-三重四级杆串联质谱测定法。
本发明任一方面或该任一方面的任一实施方案所具有的任一技术特征同样适用其它任一实施方案或其它任一方面的任一实施方案,只要它们不会相互矛盾,当然在相互之间适用时,必要的话可对相应特征作适当修饰。下面对本发明的各个方面和特点作进一步的描述。
本发明所引述的所有文献,它们的全部内容通过引用并入本文,并且如果这些文献所表达的含义与本发明不一致时,以本发明的表述为准。此外,本发明使用的各种术语和短语具有本领域技术人员公知的一般含义,即便如此,本发明仍然希望在此对这些术语和短语作更详尽的说明和解释,提及的术语和短语如有与公知含义不一致的,以本发明所表述的含义为准。
在本发明中如未另外说明,如未特别提及,涉及的%是重量/重量百分数。
附图说明
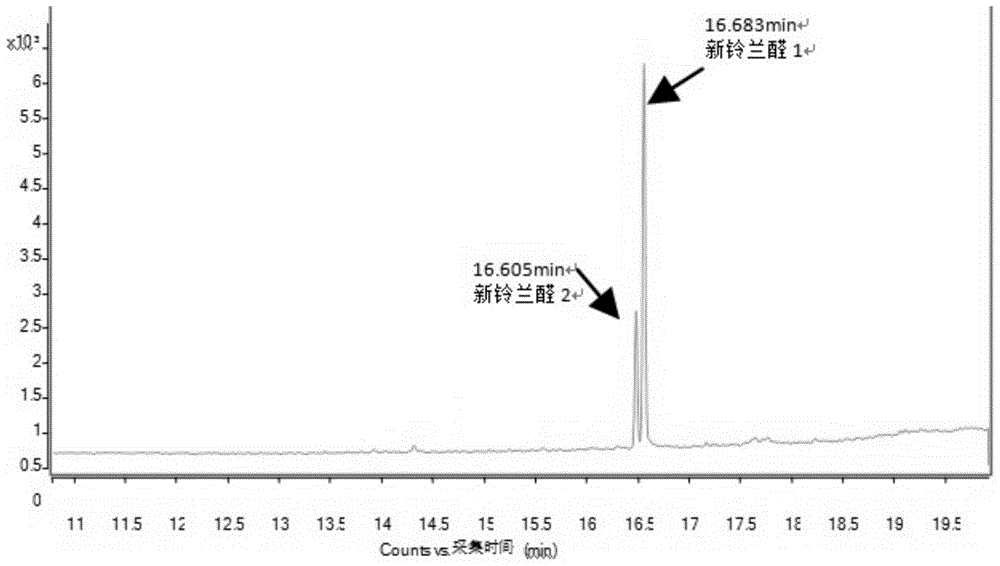
图1:新铃兰醛(含两个同分异构体)gc-ms/ms总离子流色谱图。
图2:新铃兰醛ⅰ的二级质谱图[107.1(定量离子)、77.1、79.1]。
图3:新铃兰醛ⅱ的二级质谱图[77.0(定量离子)、79.1、103.1]。
具体实施方式
通过下面的实施例可以对本发明进行进一步的描述,然而,本发明的范围并不限于下述实施例。本领域的专业人员能够理解,在不背离本发明的精神和范围的前提下,可以对本发明进行各种变化和修饰。本发明对试验中所使用到的材料以及试验方法进行一般性和/或具体的描述。虽然为实现本发明目的所使用的许多材料和操作方法是本领域公知的,但是本发明仍然在此作尽可能详细描述。
检测例1:测定各种物料中新铃兰醛含量的方法
在本发明中,如未另外说明,测定各种物料中新铃兰醛含量所用气相色谱法是气相色谱-三重四级杆串联质谱测定法,其参考cn102393438b之说明书[0016]至[0038]所述样品处理方法以及色谱条件进行,相关参数或条件作适当调整,具体方法如下:
(1)样品处理:
精密称取各种形式的样品,相当于含有新铃兰醛约10mg,将其置于50ml容量瓶中,加入甲醇约25ml,室温下超声处理20min,冷却至室温后定容至刻度,摇匀,过滤后,精密量取5ml上清液置50ml量瓶中,加甲醇稀释至刻度,摇匀;再精密量取5ml置50ml量瓶中,加甲醇稀释至刻度,摇匀,得到约2μg/ml浓度的溶液(上述配液过程中,还可以根据具体情况适当调整取样量和稀释倍数,使最终所得溶液中新铃兰醛浓度在1.5~2.5μg/ml范围内即可;或者还可以进一步稀释以达到与待检样品基本相当的浓度数量级);过0.45μm微孔ptfe滤膜后供测定。在此处理过程中,氯化钾不溶于甲醇,不会引入到气相色谱系统。使用市售新铃兰醛对照品(货号c14232000,批号:111528,购自dr.ehrenstorfergmbh)作为对照,可以用于测定本发明工艺过程中的各种物料例如新铃兰醛粗品或者新铃兰醛精馏品,亦可用于标定本发明制得的包含氯化钾的新铃兰醛对照品,接着还可以使用该经过标定的包含氯化钾的新铃兰醛对照品作为对照,用于测定本发明工艺过程中的各种物料例如新铃兰醛粗品或者新铃兰醛精馏品。不论何种物质,上述样品处理方法均适用。
(2气相色谱-三重四级杆串联质谱测定条件
a.仪器配置:agilent7000c三重串联四极杆气质联用仪(gc-ms/ms);
b.毛细管色谱柱:hp-5ms毛细管柱(30m×0.25mm,1.0μm);
c.程序升温:初始温度为60℃,保持2min后以13℃/min速率升至280℃,保持1min
d.载气:高纯氦气,流速为1.0ml/min;
e.进样口温度:280℃;进样模式为分流进样,分流比5:1;
f.检测器:三重四级杆质谱(ms/ms);
g.分析温度:离子源温度230℃,四级杆温度150℃,传输线温度280℃;
h.离子化方式:ei;电离能量:70ev;
i.监测方式:多反应监测模式(mrm);
j.碰撞气体(n2)流量:1.5ml.min-1;
k.淬灭气体(he)流量:2.25ml.min-1;
l.溶剂延迟5min。
采用本发明检测例1方法测定各种试样中的新铃兰醛,各种试样的gc色谱图与文献cn102531858b色谱图类似,主要区别是保留时间延迟到16.6min附近,容易理解,保留时间延迟是由于色谱仪器及具体色谱条件差异导致;例如在新铃兰醛对照品c14232000的gc色谱图中,两个未完全分离的主峰分别位于16.605min和16.683min处,未检出明显的其它杂质峰,两峰的峰高比约1:3,计算新铃兰醛含量时以两个主峰的总面积计;其余样品的两个主峰的保留时间亦均分别在16.60min和16.68min附近,并且峰形与c14232000的图谱类似,除了有杂质峰呈现以外(与主峰达到分离度4.0以上的分离)。
已提供的典型图谱中,图1是新铃兰醛对照品c14232000的gc-ms/ms总离子流色谱图(新铃兰醛1即新铃兰醛i,新铃兰醛2即新铃兰醛ii),图2是新铃兰醛i的二级质谱图[107.1(定量离子)、77.1、79.1],图3是新铃兰醛ii的二级质谱图[77.0(定量离子)、79.1、103.1]。
实施例1
向装有温度计、机械搅拌装置和回流冷凝器的1000ml四口烧瓶中加入539g(3.5mol)月桂烯醇和0.539g对苯二酚,加热至110℃;向所述月桂烯醇中滴加235g(4.2mol)丙烯醛,滴加6h,滴加过程中,将所述反应瓶置于冷冻槽中,保持反应瓶温度在110℃~130℃;丙烯醛滴加完毕后,继续反应1h,得到反应混合物;
将所述反应混合物用200g去离子水洗涤三次,得到765g新铃兰醛粗品(gc测得新铃兰醛含量94.74%);将所述粗品在真空度2mmhg以下进行精馏,收集120℃~125℃之间的馏分,得到612g新铃兰醛(gc测得新铃兰醛含量99.72%)。
使所得新铃兰醛馏分(约1/4量,乘余量备用)导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:200,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此混合物置于-22℃的温度下冷冻10小时,再使其回复到室温,通过摇晃使粘连的细颗粒物分散,接着将此分散的细颗粒物分装到具塞玻璃瓶中(可以根据使用需求确定每瓶的分装量;在本发明中,如未特别说明,每瓶分装量相当于新铃兰醛馏分分别为10mg、100mg、500mg、1g和10g五种规格的量,根据不同使用场合可以使用不同分装规格的瓶子),密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量,即得可用作对照品检测用的新铃兰醛(gc色谱纯度99.69%,细颗粒物中新铃兰醛的标定含量为332.04mg/g)。术语“标定含量”是指每1克物料中测定到的新铃兰醛量(以mg表示)。
实施例2
向装有温度计、机械搅拌装置和回流冷凝器的1000ml四口烧瓶中加入539g(3.5mol)月桂烯醇和0.539g对苯二酚,加热至110℃;向所述月桂烯醇中滴加216g(3.86mol)丙烯醛,滴加6h,滴加过程中,将所述反应瓶置于冷冻槽中,保持反应瓶温度在110℃~130℃;丙烯醛滴加完毕后,继续反应1h,得到反应混合物;
将所述反应混合物用180g去离子水洗涤三次,得到758g新铃兰醛粗品(gc测得新铃兰醛含量94.41%);将所述粗品在真空度2mmhg以下进行精馏,收集120℃~125℃之间的馏分,得到606g新铃兰醛(gc测得新铃兰醛含量99.69%)。
使所得新铃兰醛馏分(约1/4量,乘余量备用)导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:180,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此混合物置于-18℃的温度下冷冻12小时,再使其回复到室温,通过摇晃使粘连的细颗粒物分散,接着将此分散的细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量,即得可用作对照品检测用的新铃兰醛(gc色谱纯度99.66%,细颗粒物中新铃兰醛的标定含量为357.41mg/g)。
实施例3
向装有温度计、机械搅拌装置和回流冷凝器的1000ml四口烧瓶中加入539g(3.5mol)月桂烯醇和0.539g对苯二酚,加热至110℃;向所述月桂烯醇中滴加196g(3.5mol)丙烯醛,滴加6h,滴加过程中,将所述反应瓶置于冷冻槽中,保持反应瓶温度在110℃~130℃;丙烯醛滴加完毕后,继续反应1h,得到反应混合物;
将所述反应混合物用180g去离子水洗涤三次,得到748g新铃兰醛粗品(gc测得新铃兰醛含量88.46%);将所述粗品在真空度2mmhg以下进行精馏,收集120℃~125℃之间的馏分,得到502g新铃兰醛(gc测得新铃兰醛含量99.41%)。
使所得新铃兰醛馏分(约1/4量,乘余量备用)导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:220,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此混合物置于-25℃的温度下冷冻8小时,再使其回复到室温,通过摇晃使粘连的细颗粒物分散,接着将此分散的细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量,即得可用作对照品检测用的新铃兰醛(gc色谱纯度99.56%,细颗粒物中新铃兰醛的标定含量为312.34mg/g)。
实施例11:在室温下,将实施例1所得新铃兰醛馏分(约1/4量)导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:200,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量(gc色谱纯度99.65%,细颗粒物中新铃兰醛的标定含量为332.31mg/g)。
实施例12:在室温下,将实施例2所得新铃兰醛馏分(约1/4量)导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:180,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量(gc色谱纯度99.70%,细颗粒物中新铃兰醛的标定含量为357.21mg/g)。
实施例13:在室温下,将实施例3所得新铃兰醛馏分(约1/4量)导入预先粉碎至可通过200目筛的氯化钾粉末中,新铃兰醛与氯化钾的重量比为100:220,密塞,充分混合均匀,形成容易粘连而不能自由流动的细颗粒物,将此细颗粒物分装到具塞玻璃瓶中,密塞,采用气相色谱分析以标定此细颗粒物中新铃兰醛的含量(gc色谱纯度99.53%,细颗粒物中新铃兰醛的标定含量为312.74mg/g)。
实施例21:将实施例1所得新铃兰醛馏分(约1/20量)分装到具塞玻璃瓶中,密塞,将其置于-22℃的温度下冷冻10小时,使其回复到室温;经测定,gc色谱纯度与未冷冻时相同。
实施例22:将实施例2所得新铃兰醛馏分(约1/20量)分装到具塞玻璃瓶中,密塞,将其置于-18℃的温度下冷冻12小时,使其回复到室温;经测定,gc色谱纯度与未冷冻时相同。
实施例23:将实施例3所得新铃兰醛馏分(约1/20量)分装到具塞玻璃瓶中,密塞,将其置于-25℃的温度下冷冻8小时,使其回复到室温;经测定,gc色谱纯度与未冷冻时相同。
根据上述实施例可见,在新铃兰醛馏分与氯化钾混合前后,gc色谱纯度基本保持不变,并且细颗粒物中新铃兰醛的标定含量与新铃兰醛-氯化钾混合理论含量基本一致,这表明,新铃兰醛与氯化钾混合不会对新铃兰醛的色谱纯度和含量造成影响,仅仅相当于向油状的新铃兰醛中添加了惰性物质。
检测例2:细颗粒物的流动性测定
细颗粒物的流动性以休止角表示,休止角的测定方法参见教科书《药剂学》(奚念朱主编,第三版,人民卫生出版社出版,1996年4月第3版,isbn7-117-00026-0)第248-249页描述的方法,具体使用第249页第3-5行所述的“固定漏斗法”。
经测定,实施例1~3经冷冻处理所得分散的细颗粒物的休止角分别为51.4°、48.6°、49.8°,具有一定的流动性;实施例11~13所得细颗粒物均不能自由流动,休止角均大于70°;实施例21~23的物料为粘稠的油状物。
根据上述流动性测定结果,可知实施例1~3的产物在后续作为对照品使用或者作为其它产品的添加剂使用时均是一种非常容易让人们接受的物性。
检测例3:挥发性测定1
由于新铃兰醛是一种挥发性成分,不论其是作为检测用的对照品,还是作为其它产品的添加剂使用,挥发性都将严重影响产品应用的准确性,考察物料的挥发性将有助于了解产品的性能,为其后续使用将提供极其有意义的参考价值。
以c14232000为本试验测定用的标准对照品,将分装到玻璃瓶中的包含氯化钾的细颗粒物料(1g/瓶,实施例1~3和实施例11~13中最终所得细颗粒物)置于25℃温度下平衡12小时,测定此时的gc色谱纯度和新铃兰醛标定含量,此色谱纯度和标定含量为0月时的结果;接着使各个玻璃瓶打开密封塞,敞口,在25℃温度下放置1月,再测定各物料的gc色谱纯度及新铃兰醛标定含量;1月的gc色谱纯度和标定含量数据除以0月数据再乘以100%即为1月相应数据的相对百分数,例如1月相对色谱纯度为:1月色谱纯度除以0月色谱纯度再乘以100%所得的百分数,同样方式可得1月相对标定含量。
结果:
各试样0月结果与上文制备所得结果基本相同,例如,实施例1的0月细颗粒物中新铃兰醛gc色谱纯度99.68%、流动细颗粒物中新铃兰醛标定含量为332.02mg/g;
在1月时,实施例1~3细颗粒物的相对色谱纯度分别为98.97%、98.77%、99.16%,实施例1~3细颗粒物的相对标定含量分别为97.36%、96.81%、95.84%;实施例11~13细颗粒物的相对色谱纯度分别为96.41%、95.62%、96.30%,实施例11~13细颗粒物的相对标定含量分别为82.37%、83.61%、81.32%。
根据上述结果可见,实施例1~3细颗粒物的色谱纯度降至其0月时的98.7~99.2%,标定含量降至其0月时的95.8~97.4%,色谱纯度基本未降低,标定含量微有降低;但是未经冷冻而不能形成自由流动的实施例11~13的细颗粒物色谱纯度明显地降低至其0月时的95.6~96.5%,标定含量大大降低至其0月时的81.3~83.7%,色谱纯度有所降低,标定含量显著地大大降低。关于色谱纯度与标定含量变化不同步,其原因可能在于,细颗粒物中的挥发性组分基本上同步地挥发,而氯化钾又不会出现在gc色谱图的数据中,因此对相对色谱纯度基本没有大的影响;但是氯化钾是不挥发的,因此反映氯化钾与挥发性成分相对量的标定含量就会随着挥发性组分的挥发而产生变化,即挥发性组分挥发越多,细颗粒物中新铃兰醛标定含量就越小,相对标定含量就越低。从这个意义上讲,实施例1~3细颗粒物比之于实施例11~13的细颗粒物中新铃兰醛挥发量显著更低,新铃兰醛在细颗粒物中的稳定性显著地更好。
检测例4:挥发性测定2
照如下方式制备一些物料:在实施例1~3所得新铃兰醛精馏品中分别添加1/5重量的作为内标物质的苯甲醇,混合均匀,得到三个添加了内标物质的新铃兰醛精馏品,分别称为实施例1x、实施例2x、实施例3x样品;接着:
(a)分别照实施例1~3方法向实施例1x、实施例2x、实施例3x样品中添加氯化钾以及接着冷冻,分别得到三个可自由流动的细颗粒物,分别称为实施例1y、实施例2y、实施例3y样品;
(b)分别照实施例11~13方法向实施例1x、实施例2x、实施例3x样品中添加氯化钾但不进行冷冻,分别得到三个不可自由流动的细颗粒物,分别称为实施例11y、实施例12y、实施例13y样品;
(c)分别照实施例21~23方法对三个添加了内标物质的新铃兰醛精馏品即实施例1x、实施例2x、实施例3x样品进行冷冻,分别得到三个油状物分别称为实施例21y、实施例22y、实施例23y样品。
由于苯甲醇的挥发性相对于新铃兰醛而言非常弱,因此测定各种物料中新铃兰醛/苯甲醇含量比(更直观的是通过gc图中新铃兰醛/苯甲醇峰面积比表征),可以反映新铃兰醛在各种物料中的挥发情况。
将本检测例4所得各种试样分装到玻璃瓶中(1g/瓶)置于25℃温度下平衡12小时,测定此时的新铃兰醛/苯甲醇含量比,此含量比为0月时的结果;接着使各个玻璃瓶打开密封塞,敞口,在25℃温度下放置1月,再测定各物料的含量比,其为1月含量比;1月含量比除以0月含量比再乘以100%即为1月的相对含量比。
经测定:
实施例1x及由其制得的实施例1y、实施例11y、实施例21y四者的0月含量比均在5.216~5.228范围内,例如实施例1x含量比为5.223;实施例2x及由其制得的实施例2y、实施例12y、实施例22y四者的0月含量比均在5.054~5.068范围内,例如实施例2x含量比为5.063;实施例3x及由其制得的实施例3y、实施例13y、实施例23y四者的0月含量比均在5.178~5.191范围内,例如实施例3x含量比为5.186;
在1月时,实施例1x、实施例2x、实施例3x样品,实施例11y、实施例12y、实施例13y样品,实施例21y、实施例22y、实施例23y样品这九个样品的相对含量比均在75.4~80.3%范围内,例如实施例1x、实施例11y、实施例21y三者在1月时相对于各自0月时的相对含量比分别为78.74%、79.48%、75.93%;
在1月时,实施例1y、实施例2y、实施例3y这三个样品在1月时相对于各自0月时的相对含量比分别为98.53%、97.18%、98.34%;
这些结果表明,由同一种添加内标物的新铃兰醛经过各种处理后,新铃兰醛/苯甲醇含量比基本不变;但是这些试样在经历1个月的挥发试验后,经历了实施例1~3所示与氯化钾混合接着进行冷冻处理的试样中新铃兰醛基本未见明显的挥发,即经历与氯化钾混合接着进行冷冻处理的试样中新铃兰醛的稳定性显著提高;仅仅添加氯化钾而不进行冷冻、或者仅进行冷冻而不添加氯化钾时,均不能有效提高新铃兰醛的稳定性。
检测例5:作为对照品的应用
使用本发明呈可流动细颗粒物形式的新铃兰醛作为对照品测定化妆品中添加的新铃兰醛。
市售抽检得到的呈乳膏剂形式的防晒化妆品(产自广东某企业),疑似其中添加有新铃兰醛,使用本发明呈可流动细颗粒物形式的新铃兰醛作为对照品测定该化妆品中的新铃兰醛含量,样品处理方法如下:
称取1.0g(精确至0.001g)试样置于25ml比色管(有10ml刻度)中,加入甲醇约5ml,于超声波清洗仪上超声15min,再定容到10ml,混匀,以12000r/min高速离心15min,取上清液加入2g无水na2so4脱水,取2ml清液作为待净化液,置于通过式spe小柱(oasisprimehlb),直接收集流出液,经0.22μm有机滤膜过滤,滤液供测定用;
另分别用c14232000、本发明实施例1所得经冷冻处理的包含氯化钾和新铃兰醛的细颗粒物、本发明实施例2所得经冷冻处理的包含氯化钾和新铃兰醛的细颗粒物、本发明实施例3所得经冷冻处理的包含氯化钾和新铃兰醛的细颗粒物四者作为对照品,照检测例1方法配制对照品溶液并进行测定。在配制对照品溶液时根据待检化妆品供试品溶液中新铃兰醛含量对这些对照品溶液进行适当稀释。分别以上述四种对照品计算的上述防晒化妆品中新铃兰醛含量分别为1.163mg/g、1.166mg/g、1.159mg/g、1.162mg/g,显示使用四种对照品测定同一试样的结果基本相同。这表明,本发明实施例1~3所得能够自由流动的细颗粒物形式的新铃兰醛完全适合作为测定用的对照品。
上述内容详细描述了本发明的优选实施方式。但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何方式进行组合。为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要不违背本发明的思想,其同样应当视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!