一种荷丹片的质量检测方法与流程
本发明属于药学领域,涉及一种中药片剂的质量检测方法,特别涉及荷丹片的质量检测方法。
背景技术:
荷丹片现执行2015年中国药典标准,包括荷叶薄层鉴别、补骨脂薄层鉴别、番泻叶薄层鉴别、荷叶碱含量测定等。但荷丹片由5味中药材组成,药用物质基础复杂,且存在部分肝毒性成分,为了能够更好的控制产品的质量,提高药品的安全性,本发明根据荷丹片主要原药材的特点,制定药材的薄层鉴别方法、部分毒性成分的限量检测方法及有效成分含量的测定方法。
技术实现要素:
本发明的目的在于提供一种荷丹片的质量检测方法。
本发明所述的质量检测方法,包括对中药原料的鉴别、部分毒性成分的限量检测方法和有效成分的含量测定。
具体包括:对荷叶、补骨脂、丹参、山楂和番泻叶的鉴别,对番泻叶中的大黄酸以及对补骨脂中的补骨脂素、异补骨脂素、补骨脂苷、异补骨脂苷、荷叶中的有效成分荷叶碱、丹参中的有效成分丹酚酸b进行含量测定等步骤。
其中:
荷叶的薄层鉴别:
(1)取本品薄膜衣片,加氨试液溶解,加二氯甲烷振摇提取,二氯甲烷液浓缩,用盐酸溶液萃取,酸水液用浓氨试液调节ph值至9~10,再用二氯甲烷液振摇提取,二氯甲烷液蒸干,残渣加二氯甲烷溶解,作为供试品溶液。另取荷叶碱对照品,加二氯甲烷制成对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,分别吸取上述两种供试品溶液,分别点于同一硅胶g薄层板上,以正丁醇-醋酸-水为展开剂,展开,取出,晾干,喷以显色剂,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。
补骨脂的薄层鉴别:
(2)取本品薄膜衣片,加乙醇超声,放冷,滤过,蒸干,残渣加乙醇溶解作为供试品溶液。取补骨脂对照药材同法制得对照药材溶液。另取补骨脂素、异补骨脂素对照品,加乙醇制成混合溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述三种溶液分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯为展开剂,展开,取出,晾干,喷以显色剂,置紫外光灯下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同的两个荧光斑点。
丹参的薄层鉴别:
(3)取本品薄膜衣片,加乙醇超声,过滤,滤液挥干,残渣加乙酸乙酯溶解作为供试品溶液。取丹参对照药材同法制得对照药材溶液。另取丹参酮ⅱa对照品,加乙醇制成对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液、对照药材溶液和对照品溶液分别点于同一硅胶g薄层板上,以石油醚:乙酸乙酯为展开剂,展开,取出,晾干,在紫外光灯下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色荧光斑点。
山楂的薄层鉴别:
(4)取本品薄膜衣片,加甲醇加热回流,过滤,滤液挥干,残渣加水溶解,用乙酸乙酯振摇提取,乙酸乙酯液蒸干,残渣加甲醇溶解作为供试品溶液。另取山楂对照药材加水提取,滤过,浓缩,用乙酸乙酯振摇提取,同法制成对照药材溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液、对照药材溶液,分别点于同一硅胶g薄层板上,以环已烷:乙酸乙酯:甲酸为展开剂,展开,取出,晾干,喷以显色剂,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
番泻叶的薄层鉴别:
(5)取本品薄膜衣片,加甲醇-乙酸乙酯混合溶液加热回流,过滤,滤液蒸干,残渣加甲醇溶解作为供试品溶液。取番泻叶对照药材加甲醇加热回流,滤过,滤液蒸干,残渣加水溶解,滤过,滤液用二氯甲烷振摇提取,二氯甲烷液用碳酸钠溶液振摇提取,碳酸钠溶液用盐酸调节ph值至2,再用二氯甲烷振摇提取,二氯甲烷液用水洗涤,蒸干,残渣加甲醇溶解作为对照药材溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液与对照药材溶液分别点于同一硅胶h薄层板上,以正己烷:乙酸乙酯:甲酸为展开剂,展开,取出,晾干。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,置氨蒸气中熏后,斑点变成红色。
优选的,荷叶的薄层鉴别,步骤如下:
取本品薄膜衣片3g,研细,加氨试液20ml溶解,加二氯甲烷振摇提取3次,每次30ml,合并二氯甲烷液,浓缩至约10ml用0.01mol/l的盐酸溶液20ml萃取,弃去二氯甲烷液,酸水液用浓氨试液调节ph值至9~10,再用二氯甲烷液振摇提取3次,每次10ml,合并二氯甲烷液,蒸干,残渣加二氯甲烷1ml使溶解,作为供试品溶液。另取荷叶碱对照品,加二氯甲烷制成每1ml含0.3mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,分别吸取上述两种供试品溶液6μl、10μl,分别点于同一硅胶g薄层板上,以正丁醇-醋酸-水(4:1:1)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液,供试品色谱中,在与对照品色谱相应的位置上,显相同顏色的斑点。
优选的,补骨脂的薄层鉴别,步骤如下:
取本品薄膜衣片2g,研细,加乙醇10ml,超声15分钟,放冷,滤过,滤液蒸干,残渣加乙醇2ml使溶解,作为供试品溶液。取补骨脂对照药材粉末0.5g,同法制得对照药材溶液。另取补骨脂素、异补骨脂素对照品,加乙醇制成每1ml含2mg的混合溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述三种溶液各2~5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯(4:1)为展开剂,展开,取出,晾干,喷以10%氢氧化钾乙醇溶液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同的两个荧光斑点。
优选的,丹参的薄层鉴别,步骤如下:
取本品薄膜衣片2g,研细,加乙醇10ml,超声15分钟,过滤,滤液挥干,残渣加乙酸乙酯1ml使溶解,作为供试品溶液。取丹参对照药材粉末0.5g,同法制得对照药材溶液。另取丹参酮ⅱa对照品,加乙醇制成每1ml含1mg的溶液,作为对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液10μl、对照药材溶液和对照品溶液各5μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶g薄层板上,以石油醚(60~90℃)∶乙酸乙酯(4:1)为展开剂,展开,取出,晾干,在紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色荧光斑点。
优选的,山楂的薄层鉴别,步骤如下:
取本品薄膜衣片1.2g,研细,加甲醇50ml,加热回流30分钟,过滤,滤液挥干,残渣加水20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取山楂对照药材4g,加水100ml,提取1小时,滤过,滤液浓缩至20ml,用稀盐酸调节ph值至1~2,用乙酸乙酯振摇提取2次,同法制成对照药材溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液10μl、对照药材溶液20μl,分别点于同一硅胶g薄层板上,以环已烷-乙酸乙酯-甲酸(20:20:1)为展开剂,展开,取出,晾干,喷以2%三氯化铁乙醇溶液,在105℃加热至斑点显色清晰,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
优选的,番泻叶的薄层鉴别,步骤如下:
取本品薄膜衣片1.5g,研细,加甲醇-乙酸乙酯(1:1)的混合溶液15ml,加热回流15分钟,放冷,过滤,滤液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。取番泻叶对照药材0.5g,加甲醇15ml,加热回流15分钟,放冷,滤过,滤液蒸干,残渣加水15ml使溶解,滤过,滤液用二氯甲烷振摇提取2次,每次15ml,合并二氯甲烷液,用5%碳酸钠溶液25ml振摇提取,弃去二氯甲烷液,碳酸钠溶液用盐酸调节ph值至2,再用二氯甲烷30ml振摇提取,分取二氯甲烷液,用水洗涤2次,每次30ml,二氯甲烷液蒸干,残渣加甲醇1ml使溶解,作为对照药材溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液与对照药材溶液各10μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶h薄层板上,以正己烷-乙酸乙酯-甲酸(3:1:0.1)为展开剂,展开,取出,晾干。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,置氨蒸气中熏后,斑点变成红色。
本发明还包括对番泻叶中的大黄酸进行限量测定。
其中大黄酸限量测定方法如下:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%甲酸水溶液(31:69)为流动相;检测波长为254nm。理论板数按大黄酸峰计算应不低于5000。
对照品溶液的制备取大黄酸对照品适量,精密称定,加甲醇制成每1ml含1μg的溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声处理20分钟,取出,放至室温,并定容至刻度,摇匀,14000rpm离心10分钟,取上清液,即得。
测定方法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
本品每片含番泻叶以大黄酸(c15h18o6)计,不得过0.10mg。
本发明还包括对补骨脂中的补骨脂素、异补骨脂素、补骨脂苷、异补骨脂苷,丹参中的有效成分丹酚酸b,荷叶中的有效成分荷叶碱进行了含量测定。
其中补骨脂中的补骨脂素、异补骨脂素、补骨脂苷、异补骨脂苷的含量测定方法如下:
(1)补骨脂素、异补骨脂素
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(20:80)为流动相;检测波长为246nm,柱温60℃,流速0.3ml/min。理论板数按补骨脂素、异补骨脂素峰计算应不低于5000。
对照品溶液的制备取补骨脂素、异补骨脂素对照品适量,精密称定,分别加甲醇制成每1ml各含5μg的混合对照品溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声处理15分钟,取出,放至室温,并定容至刻度,摇匀,14000rpm离心10分钟,取上清液,即得。
测定法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
(2)补骨脂苷、异补骨脂苷
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.1%甲酸水溶液(15.7:84.3)为流动相;检测波长为246nm,柱温50℃,流速0.3ml/min。理论板数按补骨脂苷计算应不低于5000。
对照品溶液的制备取补骨脂苷、异补骨脂苷对照品适量,精密称定,加50%甲醇制成每1ml分别含12μg、8μg的混合对照品溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声处理20分钟,取出,放至室温,并定容至刻度,摇匀,取5ml置10ml容量瓶中,用10%甲醇定容至刻度,摇匀,14000rpm离心10分钟,取上清液,即得。
测定方法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
本品每片含盐补骨脂以游离型补骨脂素(c11h6o3)和异补骨脂素(c11h6o3)总量计,薄膜衣每片不得少于0.21mg;本品每片含盐补骨脂以游离型和结合型补骨脂素(c11h6o3)和异补骨脂素(c11h6o3)(补骨脂苷、异补骨脂苷与补骨脂素、异补骨脂素的折算系数0.5082)总量计,薄膜衣每片不得多于3.38mg。
其中丹参中的有效成分丹酚酸b的含量测定方法如下:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%甲酸水溶液(18:82)为流动相;检测波长为286nm,柱温50℃,流速0.3ml/min。理论板数按丹酚酸b峰计算应不低于5000。
对照品溶液的制备取丹酚酸b对照品适量,精密称定,加75%甲醇制成每1ml含29μg的溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于50ml容量瓶中,加75%甲醇适量,超声处理20分钟,取出,放置室温,并定容至刻度,摇匀,取5ml置10ml容量瓶中50%甲醇定容至刻度,混匀,14000rpm离心10分钟,取上清液,即得。
测定方法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
本品每片含丹参以丹酚酸b(c36h30o16)计,薄膜衣片不得少于1.37mg。
其中荷叶碱含量限量测定方法如下:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水-三乙胺-冰醋酸(27:70.6:1.6:0.78)为流动相;检测波长为270nm。理论板数按荷叶碱峰计算应不低于2000。
对照品溶液的制备取荷叶碱对照品适量,精密称定,加甲醇制成每1ml含16μg的溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.4g,精密称定,用加浓氨试液5ml湿润,加三氯甲烷60ml,加热回流3小时,放冷,移至分液漏斗中,分取三氯甲烷层,余留物再用三氯甲烷25ml分3次振摇提取,合并三氯甲烷液,回收三氯甲烷至干,残渣用乙醇溶解,转移至10ml量瓶中,加乙醇至刻度,滤过,取续滤液,即得。
测定方法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。
本品每片含荷叶以荷叶碱(c19h21no3)计,薄膜衣片不得少于0.25mg。
本发明所述的荷丹片的质量检测方法,是经过以下实验筛选得到的:
荷丹片鉴别项目验证研究:
1、荷叶的tlc鉴别
采用薄层色谱法,以荷叶碱对照品为鉴别荷丹片中的荷叶。
对2015版《中国药典》荷丹片中荷叶薄层鉴别方法进行优化,采用二氯甲烷作为供试品及对照药材的提取溶剂和点样溶剂,该方法使用的溶剂毒性更小、更安全、环保;增加了0.01mol/l的盐酸溶液的用量,以更完全的反萃取二氯甲烷中的荷叶碱,提高薄层灵敏度。
试验如下:
1.1实验材料:
荷叶碱对照品:由中国食品药品检定研究院提供,含量测定用。硅胶g薄层板:青岛海洋化工有限公司生产,规格10×20cm。
1.2供试品溶液的制备:
取本品薄膜衣片3g,研细,加氨试液20ml溶解,加二氯甲烷振摇提取3次,每次30ml,合并二氯甲烷液,浓缩至约10ml用0.01mol/l的盐酸溶液20ml萃取,弃去二氯甲烷液,酸水液用浓氨试液调节ph值至9~10,再用二氯甲烷液振摇提取3次,每次10ml,合并二氯甲烷液,蒸干,残渣加二氯甲烷1ml使溶解,作为供试品溶液。
1.3对照品溶液的制备:
取荷叶碱对照品,加二氯甲烷制成每1ml含0.3mg的溶液,作为对照品溶液。
1.4试验条件
展开剂:正丁醇-醋酸-水(4:1:1);
薄层板:硅胶g薄层板;
点样量:供试品溶液6μl、对照品溶液10μl;
检视方法:喷以稀碘化铋钾试液,在日光下检视。
1.5实验结果:
结果显示:供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。该项鉴别斑点清晰,操作简便,稳定可行,具有特征鉴别意义。见图1。2、补骨脂的tlc鉴别:
采用薄层色谱法,以补骨脂中的特征性成分补骨脂素和异补骨脂素为对照品、盐补骨脂对照药材鉴别荷丹片中补骨脂。
对2015版《中国药典》荷丹片中补骨脂薄层鉴别方法进行优化,采用乙醇作为供试品及对照药材的提取溶剂和点样溶剂,该方法使用低毒溶剂、更安全、环保。
试验如下:
2.1实验材料:
补骨脂素、异补骨脂素对照品:由中国食品药品检定研究院提供,供含量测定用,批号分别为110739-201617、110738-201715。
硅胶g薄层板:青岛海洋化工有限公司生产,规格10×20cm。
2.2供试品溶液的制备:
取本品薄膜衣片粉末2g,加乙醇10ml,超声15分钟,放冷,滤过,滤液蒸干,残渣加乙醇2ml使溶解,作为供试品溶液。
2.3对照品溶液的制备:
取补骨脂对照药材粉末0.5g,同法制得对照药材溶液。取阴性制剂2g,同法制的阴性对照溶液。另取补骨脂素、异补骨脂素对照品,加乙醇制成每1ml各含2mg的混合溶液,作为对照品溶液。
2.4试验条件:
展开剂:正己烷:乙酸乙酯(4:1);
薄层板:硅胶g薄层板;
点样量:缺盐补骨脂阴性制剂、补骨脂对照药材、荷丹片样品溶液各5μl,补骨脂素、异补骨脂素混合对照品溶液2μl;
检视方法:喷以10%氢氧化钾乙醇溶液,置紫外光灯(365nm)下检视。
2.5实验结果:
结果显示:样品均得到较好的分离效果,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点,且斑点显色清晰,阴性样品无干扰,说明该鉴别方法专属性好,方法可行,具有特征鉴别意义。见图2。
3、丹参的tlc鉴别:
丹参中主要含有以丹酚酸b为代表的水溶性成分和以丹参酮ⅱa为代表的脂溶性成分,因丹酚酸b作为含量测定指标列入含量测定项下,因此,荷丹片中丹参的薄层鉴别研究主要以丹参酮ⅱa作为对照品开展薄层鉴别,同时采用丹参对照药材进行对照。
试验如下:
3.1实验材料:
丹参酮ⅱa对照品:由中国食品药品检定研究院提供,批号为110766-201520。
硅胶g薄层板:由青岛海洋化工有限公司提供,规格10×20cm。
3.2供试品溶液的制备:
取本品薄膜衣片粉末2g,加乙醇10ml,超声15分钟,过滤,滤液挥干,残渣加乙酸乙酯1ml使溶解,作为供试品溶液。
3.3对照品溶液的制备:
取丹参对照药材粉末0.5g,同法制得对照药材溶液。取阴性制剂2g,同法制得阴性制剂对照溶液。另取丹参酮ⅱa对照品,加乙醇制成每1ml含1mg的溶液,作为对照品溶液。
3.4试验条件:
展开剂:石油醚(60~90℃):乙酸乙酯(4:1);
薄层板:硅胶g薄层板;
点样量:缺丹参阴性制剂、丹参对照药材、荷丹片样品溶液各10μl、丹参酮ⅱa对照品溶液5μl;
检视方法:在紫外光灯(365nm)下检视。
3.5实验结果
结果显示:样品均得到较好的分离效果,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点,且斑点显色清晰,阴性样品无干扰。说明该鉴别方法专属性好,方法可行,具有特征鉴别意义。结果见图3。4、山楂的tlc鉴别:
采用薄层色谱法,以山楂对照药材为对照鉴别荷丹片中的山楂。
试验如下:
4.1实验材料:
山楂对照药材:由中国食品药品检定研究院提供,批号为:121626-201402。
硅胶g薄层板:由青岛海洋化工有限公司提供,规格10×20cm。
4.2供试品溶液的制备:
取本品薄膜衣片1.2g,研细,加甲醇50ml,加热回流30分钟,过滤,滤液挥干,残渣加水20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加1ml乙醇溶解,作为供试品溶液。
4.3对照药材溶液的制备:
取山楂对照药材4g,加水100ml,提取1小时,滤过,滤液浓缩至20ml,用稀盐酸调节ph值至1~2,用乙酸乙酯振摇提取2次,同法制成对照药材溶液。取阴性制剂2g,同法制的阴性对照溶液。
4.4试验条件:
展开剂:环已烷-乙酸乙酯-甲酸(20:20:1);
薄层板:硅胶g薄层板;
点样量:山楂的阴性制剂、荷丹样品溶液各10μl、山楂对照药材溶液20μl;
检视方法:喷以2%三氯化铁乙醇溶液,在105℃加热至斑点显色清晰。
4.5实验结果:
结果显示:样品均得到较好的分离效果,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且斑点显色清晰,阴性样品无干扰,说明该鉴别方法专属性好,方法可行,具有特征鉴别意义。结果见图4。
5.番泻叶的tlc鉴别:
采用薄层色谱法,以番泻叶对照药材为对照鉴别荷丹片中的番泻叶。
试验如下:
5.1实验材料:
番泻叶对照药材:由中国食品药品检定研究院提供,批号:120996-201205。
硅胶g薄层板:由青岛海洋化工有限公司生产,规格10×20cm。
5.2供试品液的制备:
取本品薄膜衣片1.5g,研细,加甲醇-乙酸乙酯(1:1)的混合溶液15ml,加热回流15分钟,放冷,过滤,滤液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。
5.3对照药材溶液的制备:
取番泻叶对照药材1g,加入10ml乙醇,超声提取30min,离心,取上清液,蒸干,残渣加水10ml,用60~90℃石油醚萃取3次,每次15ml,取水液蒸干,加入乙醇5ml溶解作为对照药材供试品溶液。
5.4实验条件:
展开剂:正己烷-乙酸乙酯-甲酸(3:1:0.1);
薄层板:硅胶g薄层板;
点样量:番泻叶对照药材供试品溶液5μl,其余各供试品溶液各10μl;
检视方法:置饱和氨气中熏蒸(置于密闭展开槽中)后,置254nm紫外灯下检视。
5.5实验结果
结果显示:样品均得到较好的分离效果,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且斑点显色清晰,阴性样品无干扰,说明该鉴别方法专属性好,方法可行,具有特征鉴别意义。结果见图5。
二、荷丹片限量测定项目验证研究:
荷丹片中部分成分如大黄酸(来自番泻叶)存在毒性风险。因此需进一步制定上述成分含量的安全上限,从而确保制剂的临床安全性。
现对限量测定项进行验证:
1、荷丹片中大黄酸的限量测定方法验证研究:
仪器与试药:超高效液相色谱仪:watersacquityhplc-hclass(美国沃特世公司),pda检测器;色谱柱:acquity
1)供试品溶液制备方法的条件优化
在开展丹荷丹片中大黄酸的含量测定时,系统考察了提取溶剂、溶剂体积、提取时间对荷丹片中大黄酸提取效果的影响,优选了最佳的供试品溶液制备方法。
提取溶剂考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,加入不同提取溶剂(25%甲醇、50%甲醇、75%甲醇、甲醇)适量,超声处理20min,取出后放至室温,用相应溶剂定容至刻度,摇匀,取上清液14000rpm离心10min,取上清液,进样分析。平行处理两份,取平均值。结果见表1,根据大黄酸的性质和提取率,最后确定用甲醇作为提取溶剂。
提取溶剂的体积考察
分别取荷丹片粉末1g、0.5g、0.5g,精密称定,对应置于不同体积(25ml、25ml、50ml)容量瓶中,分别加入甲醇适量,超声处理20min,取出后放至室温,用甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液进样分析。平行处理两份,取平均值。结果见表2,根据大黄酸的性质,综合考虑料液比和提取率,确定取粉末0.5g,用甲醇25ml超声提取。
提取时间考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,分别加入甲醇适量,分别超声处理10min、15min、20min,25min后取出,放至室温,用甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液进样分析。平行处理两份,取平均值。结果见表3,根据大黄酸的性质,综合考虑料液比和提取率,确定用纯甲醇25ml超声提取20min。
综上所述,同时考虑到大黄酸的性质,确定供试品溶液的制备方法:取装量差异项下的本品24片,粉碎,取0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声提取20min,取出后放至室温,并定容至刻度,摇匀,14000rpm离心10min,取上清液进样分析。
表1不同提取溶剂下大黄酸的结果分析(n=2,μg/g)
表2不同提取溶剂(甲醇)体积大黄酸的结果分析(n=2,μg/g)
表3不同提取时间大黄酸的结果分析(n=2,μg/g)
2)色谱条件
采用色谱柱acquity
3)线性关系考察
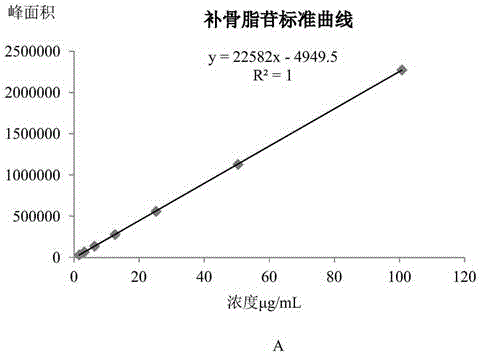
精密称取大黄酸标准品10.12mg,置于50ml容量瓶中,加二甲基亚砜适量使溶解,加甲醇定容至刻度,摇匀既得大黄酸对照品溶液(202.4μg/ml)。精密移取大黄酸对照品溶液1ml置于25ml容量瓶中,用甲醇定容,摇匀,配制成浓度分别为8.096μg/ml的对照品溶液。用甲醇逐级稀释2、4、8、16、32倍,得到一系列不同浓度的混合对照品溶液,按照色谱条件取2μl注入液相色谱仪进行检测,平行分析2次,以大黄酸的浓度x为横坐标,以大黄酸峰面积y为纵坐标绘制标准曲线;以信噪比s/n=3为检测限(lod)、s/n=10为定量限(loq)测定大黄酸的检测线和定量限。结果表明:在大黄酸的浓度范围内线性关系良好,线性结果见图7,表4。
表4大黄酸的线性回归方程、线性范围、lod、loq
4)精密度实验
日内精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次。结果见表5。
日间精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,连续进样三天,每天处理同一批号样品制备供试品溶液。结果见表6。
表5样品中大黄酸测定的日内精密度试验结果(n=6,峰面积)
表6样品中大黄酸测定的日间精密度试验结果(n=3,峰面积)
5)重复性试验
取荷丹片粉末0.5g,精密称定,称取6份,分别置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析。结果见表7。
表7样品中大黄酸测定的重复性试验结果(n=6,μg/g)
6)稳定性试验
取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,分别于0、2、4、6、8、10、12h进样分析。结果见表8。
表8样品中大黄酸测定的稳定性试验结果(n=7,峰面积)
7)加样回收试验
称取荷丹片样品0.25g,精密称定,称取6份,分别置于25ml容量瓶中,精密加入大黄酸混合对照品溶液2ml(8.096μg/ml),加甲醇适量,超声提取20min后取出,放至室温,甲醇定容,摇匀,14000rpm离心10min,取上清液既得供试品溶液。按照所述色谱条件分析,计算大黄酸加样回收率,结果见表9。
表9样品中大黄酸加样回收率试验结果(n=6)
研究结果表明,大黄酸的平均加样回收率为99.74%,rsd为2.66%。
8)16批荷丹片中大黄酸的含量测定
取装量差异项的本品24片,研细,取0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液制备方法制备,按照所述色谱条件分析,分别测定16批次的荷丹片样品中大黄酸的含量,结果见表10。
表1016批次荷丹片中大黄酸含量测定结果(n=2)
注:每片重量按0.73g计。
9)含量限定的制定
根据大生产16个批次荷丹片中大黄酸的测定结果,同时,考虑到大生产中投料药物和生产工艺的波动,将16批荷丹片大黄酸测定结果的平均值上浮50%作为上限,16批荷丹片大黄酸测定结果的平均值为0.06739mg/片,则上限为0.06739mg/片×150%=0.1011mg/片,暂定每片含番泻叶以大黄酸(c15h18o6)计,不得过0.10mg。
三、荷丹片含量测定项目验证研究:
荷丹片处方中盐补骨脂是治疗高血脂的重要药味,补骨脂素和异补骨脂素既是有效成分,也存在肝毒性的安全性风险,而且补骨脂苷和异补骨脂苷在肠道菌群作用下转化为补骨脂素和异补骨脂素,因此有必要建立其含量测定方法并制定含量的上下限;荷叶和丹参占据较重要的位置,因此对荷叶中的有效成分荷叶碱和丹参中的有效成分丹酚酸b进行了含量测定,以确保制剂的有效性。现对各含量测定项进行验证:
1、荷丹片中补骨脂素和异补骨脂素、补骨脂苷和异补骨脂苷的限量测定方法验证研究:
(异)补骨脂苷是盐补骨脂中主要成分,(异)补骨脂苷在人体肠道中反应生成(异)补骨脂素,而高浓度的(异)补骨脂素易导致肝脏损害,因此应确定“转化态”补骨脂素、异补骨脂素和“原生态”补骨脂素、异补骨脂素(荷丹片中游离型补骨脂素、异补骨脂素)总量的上、下限标准。
仪器与试药:超高效液相色谱仪:watersacquityhplc-hclass(美国沃特世公司),pda检测器;色谱柱:acquity
1)供试品溶液制备方法的条件优化
在开展丹荷丹片中补骨脂苷和异补骨脂苷的含量测定时,系统考察了提取溶剂、溶剂体积、提取时间对荷丹片中补骨脂苷、异补骨脂苷提取效果的影响,优选了最佳的供试品溶液制备方法。
提取溶剂考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,加入不同提取溶剂(25%甲醇、50%甲醇、75%甲醇、甲醇)适量,超声处理20min,取出后放至室温,用相应溶剂定容至刻度,摇匀,取上清液14000rpm离心10min,精密移取上清液5ml至10ml容量瓶中,10%甲醇定容,混匀后进样分析。平行处理两份,取平均值。结果见表11,根据补骨脂苷和异补骨脂苷的性质和提取率,最后确定用甲醇作为提取溶剂。
提取溶剂的体积考察:分别取荷丹片粉末1g、0.5g、0.5g,精密称定,对应置于不同体积(25ml、25ml、50ml)容量瓶中,分别加入甲醇适量,超声处理20min,取出后放至室温,用甲醇定容至刻度,摇匀,取上清液14000rpm离心10min,精密移取上清液5ml至10ml容量瓶中,10%甲醇定容,混匀后进样分析。平行处理两份,取平均值。结果见表12,根据补骨脂苷和异补骨脂苷的性质,综合考虑料液比和提取率,确定取荷丹片粉末0.5g,用甲醇25ml超声提取。
提取时间考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,分别加入甲醇适量,分别超声处理10min、15min、20min,25min后取出,放至室温,用甲醇定容至刻度,摇匀,取上清液14000rpm离心10min,精密移取上清液5ml至10ml容量瓶中,10%甲醇定容,混匀后进样分析。平行处理两份,取平均值。结果见表13,根据补骨脂苷和异补骨脂苷的性质,综合考虑料液比和提取率,确定用纯甲醇25ml超声提取20min。
综上所述,同时考虑到补骨脂苷和异补骨脂苷的性质,确定供试品溶液的制备方法:取本品24片,粉碎,取0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声提取20min,取出后放至室温,并定容至刻度,摇匀,取上清液14000rpm离心10min,用移液管精密移取上述溶液5ml至10ml容量瓶中,10%甲醇定容至刻度,摇匀,进样分析。
表11不同提取溶剂下补骨脂苷和异补骨脂苷的结果分析(n=2,μg/g)
表12不同提取溶剂(甲醇)体积补骨脂苷和异补骨脂苷的结果分析(n=2,μg/g)
表13不同提取时间补骨脂苷和异补骨脂苷的结果分析(n=2,μg/g)
2)色谱条件
采用色谱柱acquity
因为荷丹片中含有较多的小极性成分(如丹参中的丹参酮类成分、补骨脂中的补骨脂酚等成分)在色谱柱上有较强保留,为了减少残留成分对后续分析的影响,在完成补骨脂苷与异补骨脂苷的分析(16min)后,采用梯度洗脱的程序(18-21min,90%甲醇;21-22min,90%~15.7%甲醇;22-26min,15.7%甲醇)冲洗色谱柱,其目的主要是为了洗脱色谱柱上的强保留小极性成分,避免对后续分析的影响。
3)线性关系考察
精密称取标准品补骨脂苷20.15mg、异补骨脂苷21.20mg,置于10ml容量瓶中,加甲醇溶解,定容至刻度摇匀,得补骨脂苷对照品母液(2.015mg/ml)和异补骨脂苷对照品母液(2.120mg/ml)。精密移取补骨脂苷、异补骨脂苷对照品溶液各1ml分别置于10ml容量瓶中,用50%甲醇定容,摇匀,配制成浓度分别为:补骨脂苷0.2015mg/ml、异补骨脂苷0.2120mg/ml溶液、精密移取补骨脂苷溶液5ml和异补骨脂苷母液3ml到10ml容量瓶中,加50%甲醇定容至刻度摇匀,既得混合对照品溶液,浓度分别为100.75μg/ml、63.60μg/ml。用50%甲醇逐级稀释2、4、8、16、32、64倍,得到一系列不同浓度的混合对照品溶液,按照色谱条件取2μl注入液相色谱仪进行检测,平行分析2次,以补骨脂苷、异补骨脂苷的浓度x为横坐标,以补骨脂苷、异补骨脂苷峰面积y为纵坐标绘制标准曲线;以信噪比s/n=3为检测限(lod)、s/n=10为定量限(loq)测定补骨脂苷、异补骨脂苷的检测线和定量限。结果表明:在补骨脂苷、异补骨脂苷的浓度范围内线性关系良好,线性结果见图9,表14。
表14补骨脂苷和异补骨脂苷线性回归方程、线性范围、lod、loq
4)精密度实验
日内精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次。结果见表15。
日间精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,连续进样三天,每天处理同一批号样品制备供试品溶液,结果见表16。
表15补骨脂苷和异补骨脂苷测定的日内精密度试验结果(n=6,峰面积)
表16补骨脂苷和异补骨脂苷测定的日间精密度试验结果(n=3,峰面积)
5)重复性试验
取荷丹片粉末0.5g,精密称定,称取6份,分别置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析。结果见表17。
表17样品中补骨脂苷和异补骨脂苷测定的重复性试验结果(n=6,μg/g)
6)稳定性试验
取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,分别于0、2、4、6、8、10、12h进样分析。结果见表18。
表18样品中补骨脂苷和异补骨脂苷测定的稳定性试验结果(n=7,峰面积)
7)加样回收率试验
称取荷丹片样品0.25g六份,精密称定,称取6份,分别置于25ml容量瓶中,精密加入补骨脂苷、异补骨脂苷混合对照品溶液3ml(补骨脂苷0.1169mg/ml、异补骨脂苷0.0636mg/ml),加甲醇适量,超声提取20min后取出,放至室温,甲醇定容,摇匀,取上清液14000rpm离心10min,精密移取上清液5ml至10ml容量瓶中,10%甲醇定容,摇匀。按照所述色谱条件分析,计算补骨脂苷、异补骨脂苷加样回收率,结果见表19。
表19样品中补骨脂苷和异补骨脂苷加样回收率试验结果(n=6)
研究结果表明,补骨脂苷和异补骨脂苷的平均加样回收率分别为97.94%、97.92%,rsd分别为0.89%、0.74%。
8)16批荷丹片中补骨脂苷和异补骨脂苷总含量测定
取本品24片,研细,取0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液制备方法制备,按照所述色谱条件分析,分别测定16批次的荷丹片样品中补骨脂苷和异补骨脂苷含量总量,结果见表20。
表2016批荷丹片中补骨脂苷和异补骨脂苷总量测定结果(n=2)
注:每片重量按0.73g计。
补骨脂素和异补骨脂素参照高效液相色谱法(中国药典2015版四部通则0512)测定
仪器与试药:超高效液相色谱仪:watersacquityhplc-hclass(美国沃特世公司),pda检测器;色谱柱:acquity
1)供试品溶液制备方法的条件优化
在开展丹荷丹片中补骨脂素和异补骨脂素的含量测定时,系统考察了提取溶剂、溶剂体积、提取时间对荷丹片中补骨脂素、异补骨脂素提取效果的影响,优选了最佳的供试品溶液制备方法。
提取溶剂考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,加入不同提取溶剂(25%甲醇、50%甲醇、75%甲醇、甲醇)适量,超声处理20min,取出后放至室温,用相应溶剂定容至刻度,摇匀,取上清液14000rpm离心10min,取上清液,进样分析。平行处理两份,取平均值。结果见表21,提取溶剂选择100%甲醇,此条件下补骨脂素、异补骨脂素的提取率最高。
提取料液比考察
分别取荷丹片粉末1g、0.5g、0.5g,精密称定,对应置于不同体积(25ml、25ml、50ml)容量瓶中,分别加入甲醇适量,超声处理20min,取出后放至室温,用甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液进样分析。平行处理两份,取平均值。结果见表22,提取效率随提取料液比而增加,考虑到节省提取溶剂,料液比选为1:50较为合适,即取粉末0.5g,加25ml甲醇提取。
提取时间考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,分别加入甲醇适量,分别超声处理10min、15min、20min,25min后取出,放至室温,用甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液进样分析。平行处理两份,取平均值。结果见表23。结果表明提取效率随提取时间的延长变化不大,最后综合考虑,选择15min作为最佳提取时间。
综上所述,同时考虑到补骨脂素和异补骨脂素的性质,最后确定的最优提取方法为:取本品24片,粉碎,取0.5g,精密称定,置于25ml容量瓶中,加入适量甲醇,超声提取15min,取出后放置室温,用甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液,即得供试品溶液。
表21不同提取溶剂下补骨脂素和异补骨脂素的结果分析(n=2,μg/g)
表22不同提取溶剂(甲醇)体积补骨脂素和异补骨脂素的结果分析(n=2,μg/g)
表23不同提取时间补骨脂素和异补骨脂素的结果分析(n=2,μg/g)
2)色谱条件
采用色谱柱acquity
结果表明:采用acquity
3)线性关系考察
精密称取标准品补骨脂素11.03mg、异补骨脂素11.39mg,置于10ml容量瓶中,加甲醇溶解,定容至刻度摇匀,得补骨脂素对照品母液(1.103mg/ml)和异补骨脂素对照品母液(1.139mg/ml)。精密移取补骨脂素、异补骨脂素对照品溶液1ml置于25ml容量瓶中,用甲醇定容,摇匀,配制成浓度分别为:补骨脂素44.12μg/ml、异补骨脂素45.56μg/ml的混合对照品溶液。用甲醇逐级稀释2、4、8、16、32倍,得到一系列不同浓度的混合对照品溶液,按照色谱条件取2μl注入液相色谱仪进行检测,平行分析2次,以补骨脂素、异补骨脂素的浓度x为横坐标,以补骨脂素、异补骨脂素峰面积y为纵坐标绘制标准曲线;以信噪比s/n=3为检测限(lod)、s/n=10为定量限(loq)测定补骨脂素、异补骨脂素的检测线和定量限。结果表明:在补骨脂素、异补骨脂素的浓度范围内线性关系良好,线性结果见图11,表24。
表24补骨脂素和异补骨脂素的线性回归方程、线性范围、lod、loq
4)精密度实验
日内精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,测定其峰面积,结果见表25。
日间精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,连续进样三天,每天处理同一批号样品制备供试品溶液,测定其峰面积,结果见表26。
表25补骨脂素和异补骨脂素测定的日内精密度试验结果(n=6,峰面积)
表26补骨脂素和异补骨脂素测定的日间精密度试验结果(n=3,峰面积)
5)重复性试验
取荷丹片粉末0.5g,精密称定,称取6份,分别置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,计算荷丹片中补骨脂素、异补骨脂素的含量,重复性结果见表27。
表27样品中补骨脂素和异补骨脂素测定的重复性试验结果(n=6,μg/g)
6)稳定性试验
取荷丹片粉末0.5g,精密称定,称取6份,分别置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,分别于0、2、4、6、8、10、12h的时间间隔进行检测,结果见表28。
表28样品中补骨脂素和异补骨脂素测定的稳定性试验结果(n=7,μg/g)
7)加样回收率试验
称取荷丹片样品0.25g六份,精密称定,分别置于25ml容量瓶中,精密加入补骨脂素、异补骨脂素混合对照品溶液2ml(补骨脂素0.0441mg/ml、异补骨脂素0.0456mg/ml),按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,计算补骨脂素、异补骨脂素加样回收率,结果见表29。
表29样品中补骨脂素和异补骨脂素加样回收率试验结果(n=6)
研究结果表明,补骨脂素和异补骨脂素的平均回收率为100.46%和99.18%,rsd分别为1.72%和1.63%。
8)多批次荷丹片中补骨脂素和异补骨脂素的含量测定
取本品24片,研细,取0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液制备方法制备,按照所述色谱条件分析,分别测定16批次的荷丹片样品中补骨脂素和异补骨脂素含量总和,结果见表30。
表3016批荷丹片中补骨脂素和异补骨脂素总和测定结果(n=2)
注:每片重量按0.73g计。
9)补骨脂素和异补骨脂素、补骨脂苷、异补骨脂苷含量限度的制定
根据16个批次荷丹片中补骨脂素、异补骨脂素的测定结果,同时,考虑到大生产中投料药物和生产工艺的波动,将16批荷丹片补骨脂素、异补骨脂素测定总量的平均值下浮50%作为下限,16批荷丹片游离型补骨脂素、异补骨脂素测定总量的平均值为0.4185mg/片,则下限为0.4185mg/片×50%=0.2092mg/片,暂定每片含盐补骨脂以游离型补骨脂素(c11h6o3)和异补骨脂素(c11h6o3)的总量计,不得少于0.21mg。
将16批荷丹片游离型补骨脂素、异补骨脂素和结合型补骨脂素、异补骨脂素测定总量的平均值上浮50%作为上限,16批荷丹片游离型补骨脂素、异补骨脂素和结合型补骨脂素、异补骨脂素测定总量的平均值为2.252mg/片,则上限为2.252mg/片×150%=3.378mg/片,暂定每片含盐补骨脂以游离型补骨脂素(c11h6o3)、异补骨脂素(c11h6o3)和结合型补骨脂素(c11h6o3)、异补骨脂素(c11h6o3)(补骨脂苷、异补骨脂苷与补骨脂素、异补骨脂素的折算系数0.5082)的总量计,不得多于3.38mg。
2、荷丹片中丹酚酸b含量测定方法验证研究:
仪器与试药:超高效液相色谱仪:watersacquityhplc-hclass(美国沃特世公司),pda检测器;色谱柱:acquity
1)供试品溶液制备方法的条件优化
在开展荷丹片中丹酚酸b的含量测定时,系统考察了提取溶剂、提取料液比、提取时间三个方面对荷丹片中丹酚酸b提取效果的影响,优选了最佳的供试品溶液制备方法。
提取溶剂考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,加入不同提取溶剂(25%甲醇、50%甲醇、75%甲醇、甲醇)适量,超声处理20min,取出后放至室温,用相应溶剂定容至刻度,摇匀,14000rpm离心10min,取上清液5ml至10ml容量瓶中,用50%甲醇定容至刻度,摇匀,进样分析。平行处理两份,取平均值。结果见表31,提取溶剂选择75%甲醇水,此条件下丹酚酸b的提取率最高。
提取料液比考察:分别取荷丹片粉末1g、0.5g、0.5g,精密称定,对应置于不同体积(25ml、25ml、50ml)容量瓶中,分别加入75%甲醇适量,超声处理20min,取出后放至室温,用甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液5ml至10ml容量瓶中,用50%甲醇定容至刻度,摇匀,进样分析。平行处理两份,取平均值。结果见表32,提取效率随提取料液比而增加,考虑到节省提取溶剂,料液比选为1:50较为合适,即取粉末0.5g,加25ml75%甲醇提取。
提取时间考察:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,分别加入甲醇适量,分别超声处理10min、15min、20min,25min后取出,放至室温,用75%甲醇定容至刻度,摇匀,14000rpm离心10min,取上清液5ml至10ml容量瓶中,用50%甲醇定容至刻度,摇匀,进样分析。平行处理两份,取平均值。结果见表33,提取效率随提取时间的延长而增加,但增幅较小,最后综合考虑选择20min作为最佳提取时间。
综上所述,同时考虑到丹酚酸b的性质,确定供试品溶液的制备方法为:取装量差异项下的本品24片,粉碎,取0.5g,精密称定,置于25ml容量瓶中,加75%甲醇适量,超声提取20min,取出后放至室温,并定容至刻度,摇匀,取5ml至10ml容量瓶中,50%甲醇定容至刻度,混匀,14000rpm离心10min取上清液,即得供试品溶液。
表31不同提取溶剂下丹酚酸b的结果分析(n=2,mg/g)
表32不同提取溶剂(甲醇)体积丹酚酸b的结果分析(n=2,mg/g)
表33不同提取时间丹酚酸b的结果分析(n=2,mg/g)
2)色谱条件
采用色谱柱acquity
结果表明:采用acquity
3)线性关系考察
精密称取丹酚酸b标准品(纯度94.1%)23.31mg,置于10ml容量瓶中,加甲醇溶解,定容至刻度,摇匀既得丹酚酸b对照品溶液(2.193mg/ml)。精密移取丹酚酸b对照品溶液1ml至10ml容量瓶中,用甲醇定容,摇匀,配制成浓度分别为219.3μg/ml的对照品溶液。用甲醇逐级稀释2、4、8、16、32倍,得到一系列不同浓度的混合对照品溶液,按照色谱条件取2μl注入液相色谱仪进行检测,平行分析2次,以丹酚酸b的浓度x为横坐标,以丹酚酸b峰面积y为纵坐标绘制标准曲线;以信噪比s/n=3为检测限(lod)、s/n=10为定量限(loq)测定丹酚酸b的检测线和定量限。结果表明:在丹酚酸b的浓度范围内线性关系良好,线性结果见图13,表34。
表34丹酚酸b的线性回归方程、线性范围、lod、loq
4)精密度实验
日内精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,测定其峰面积,结果见表35。
日间精密度:取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,重复进样6次,连续进样三天,测定其峰面积,结果见表36。
表35样品中丹酚酸b测定的日内精密度试验结果(n=6,峰面积)
表36样品中丹酚酸b测定的日间精密度试验结果(n=3,峰面积)
5)重复性试验
取荷丹片粉末0.5g,精密称定,称取6份,分别置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,,重复性结果见表37。
表37样品中丹酚酸b测定的重复性试验结果(n=6,mg/g)
6)稳定性试验
取荷丹片粉末0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液的制备方法制备,按照所述色谱条件分析,分别于0、2、4、6、8、10、12h的时间间隔进行分析,结果见表38。
表38样品中丹酚酸b测定的稳定性试验结果(n=7,mg/g)
7)加样回收率试验
称取荷丹片样品0.25g六份,精密称定,分别置于25ml容量瓶中,精密加入丹酚酸b对照品溶液5ml(丹酚酸b浓度为0.1316mg/g),按照制备方法制备,按照所述色谱条件进行检测分析,结果见表39。
表39样品中丹酚酸b加样回收率试验结果(n=6)
实验结果表明,丹酚酸b的平均回收率为101.16%,rsd为2.15%,符合相关方法学的要求,可用于丹酚酸b的含量测定。
8)16批荷丹片中丹酚酸b的含量测定
取装量差异项的本品24片,研细,取0.5g,精密称定,置于25ml容量瓶中,按照所述供试品溶液制备方法制备,按照所述色谱条件分析,分别测定16批次的荷丹片样品中丹酚酸b的含量,结果见表40。
表4016批次荷丹片中丹酚酸b含量测定结果(n=2)
注:每片重量按0.73g计。
9)丹酚酸b含量限度的制定
荷丹片中的丹酚酸b有较强的抗氧化及降血脂的功效,是荷丹片的主要药效物质,因此需要进一步制定丹酚酸b的含量下限。
根据大生产16个批次荷丹片中丹酚酸b的测定结果,同时,考虑到大生产中投料药物和生产工艺的波动,将16批荷丹片丹酚酸b测定结果的平均值下浮50%作为下限,16批荷丹片丹酚酸b测定结果的平均值为2.731mg/片,则下限为2.731mg/片×50%=1.366mg/片,暂定每片含丹参以丹酚酸b(c36h30o16)计,不得少于1.37mg。
3、荷丹片中荷叶碱的含量测定方法验证研究:
1.1仪器与试药
仪器:岛津高效液相色谱仪(lc-20at),电子天平ab204-s。
试剂:甲醇为色谱纯,水为重蒸水;其他试剂均为分析纯。
对照品:荷叶碱对照品为中国食品药品检定研究院提供(110736-201725供含量测定用)。
样品:荷丹片(南昌济顺制药有限公司提供);
1.2色谱条件与系统适用性试验
色谱柱:shimadzuvp-ods(5um,250mmdzuvp);十八烷基键合硅胶为填充剂;流动相:乙腈-水-三乙胺-冰醋酸(27:70.6:1.6:0.78);流速:1ml/min;柱温:25℃;波长:270nm。
供试品溶液的制备:取本品薄膜衣片24片,研细,取约0.4g,精密称定,加浓氨试液5ml湿润,加三氯甲烷60ml,加热回流3小时,放冷,移至分液漏斗中,分取三氯甲烷层,余留物再用三氯甲烷25ml分3次振摇提取,合并氯仿三氯甲烷液,回收三氯甲烷至干,残渣用乙醇溶解,转移至10ml量瓶中,加乙醇至刻度,滤过,取续滤液,即得。
1.3线性关系的考察
精密称取在五氧化二磷干燥器中减压干燥至恒重的荷叶碱1.62mg,置100ml容量瓶中,加水溶解,并稀释至刻度,摇匀,即得。分别进样1μl、5μl、10μl、15μl、20μl、25μl,测定其峰面积,绘制标准曲线,结果见表41。
表41荷叶碱对照品测定结果
峰面积-进样量标准曲线见图14。
回归方程y=1681202.7x-4684.9r=0.99999。结果表明进样量在0.0162-0.405μg范围内线性关系良好。
1.4精密度试验
吸取对照品溶液(16.2μg/ml),重复进样5次,计算峰面积积分值的相对标准偏差,结果见表42。
表42精密度试验结果
试验结果表明:仪器的精密度良好。
1.5稳定性实验
取本品0.4g,按“供试品溶液的制备”试方法制备供试品溶液,分别于0小时、3小时、6小时、9小时、12小时、15小时,进样,测其含量。结果见表43。
表43稳定性试验结果
对照品浓度:16.2μg/ml,对照品峰面积:1141552
结果表明:供试品溶液在15小时内稳定性良好。
1.6重现性实验
取同一批号的荷丹片(180601)的样品5份,分别按拟订方法制备、测定,结果荷叶碱含量见表44。
表44重现性试验结果
对照品浓度:16.2μg/ml,对照品峰面积:1141552
结果表明:试验方法的重现性良好。
1.7加样回收率试验
(采用加样回收法)取已知含量的荷丹片(批号:180603含量0.394mg/g),精密称取0.5g,粉碎后置20ml量瓶中,加入荷叶碱对照品溶液(16.2μg/ml10ml,按“供试品溶液的制备”试方法制备,进样,计算回收率,结果见表45。
计算公式:回收率(%)=(实测量-供试品所含被测成分量)/加入对照品的量
表45回收率试验结果
对照品浓度:16.2μg/ml,对照品峰面积1144969
结果表明:本次实验的平均回收率为98.23%,rsd为2.11%,加样回收良好。
1.8样品测定
取三批样品,按质量标准中的方法测定含量,结果见表46。
荷叶碱含量以外标峰面积法计算,含量计算公式如下:
荷叶碱(mg/ml)=(a1×m2×mg/m)/(a2×m)
a1:样品峰面积a2:对照品峰面积c2:对照品浓度m:取样量
注:对照品浓度:16.2μg/ml,对照品峰面积:1120972
表46样品中荷叶碱含量测定结果
结果表明:荷丹片中荷叶碱的含量测定方法可行,本品每片(0.73g)含荷叶以荷叶碱(c19h21no3)计,不得少于0.25mg。
本发明对荷丹片质量标准5个鉴别项、1个限量测定项及4个含量检测项进行了方法学验证,结果显示荷丹片质量检测方法中的各项目操作简单、重现性好、稳定性好、检测效果准确度高,可有效的控制荷丹片的质量。同时,填补了该产品定量控制的空白,使得对该产品能够进行更有效的质量分析,更全面反映产品的质量状况,保证该产品的质量稳定性。
附图说明
图1、荷丹片中荷叶鉴别tlc图
1.缺荷叶阴性制剂2.荷叶碱对照品
3-18.16批次荷丹片样品(20180201、20180202、20180203、20180204、20180301、20180302、20190101、20190102、20190103、20190104、20190105、20190106、20190203、20190204、20190301、20190302)
图2荷丹片中补骨脂tlc图
1.缺补骨脂阴性制剂2.补(异)骨脂素对照品3.补骨脂对照药材
4-19.16批荷丹片样品(20180201、20180202、20180203、20180204、20180301、20180302、20190101、20190102、20190103、20190104、20190105、20190106、20190203、20190204、20190301、20190302)
图3荷丹片中丹参鉴别tlc图
1.缺丹参阴性制剂2.丹参酮iia对照品3.丹参对照药材
4-19.16批次荷丹片样品(20180201、20180202、20180203、20180204、20180301、20180302、20190101、20190102、20190103、20190104、20190105、20190106、20190203、20190204、20190301、20190302)
图4荷丹片中山楂鉴别tlc图
1.山楂对照药材,2-17.16批次荷丹片样品(20180201、20180202、20180203、20180204、20180301、20180302、20190101、20190102、20190103、20190104、20190105、20190106、20190203、20190204、20190301、20190302)18.缺山楂阴性制剂
图5荷丹片中番泻叶鉴别tlc图
1.番泻叶对照药材2-17.16批次荷丹片样品(20180201、20180202、20180203、20180204、20180301、20180302、20190101、20190102、20190103、20190104、20190105、20190106、20190203、20190204、20190301、20190302)18.缺番泻叶阴性制剂
图6荷丹片中大黄酸分析的色谱图
图7大黄酸对照品溶液的标准曲线
图8荷丹片中补骨脂苷和异补骨脂苷分析的色谱图
图9补骨脂苷、异补骨脂苷对照品溶液的标准曲线
图10荷丹片中补骨脂素和异补骨脂素分析的色谱图
图11补骨脂素、异补骨脂素对照品溶液的标准曲线
图12荷丹片中丹酚酸b分析的色谱图
图13丹酚酸b对照品溶液的标准曲线
图14荷叶碱对照品溶液的标准曲线
具体实施方式
通过以下具体实施例对本发明作进一步的说明,但不作为本发明的限制。
实施例1、荷丹片的质量检测
【鉴别】
1)荷叶的鉴别:取本品薄膜衣片3g,研细,加氨试液20ml溶解,加二氯甲烷振摇提取3次,每次30ml,合并二氯甲烷液,浓缩至约10ml用0.01mol/l的盐酸溶液20ml萃取,弃去二氯甲烷液,酸水液用浓氨试液调节ph值至9~10,再用二氯甲烷液振摇提取3次,每次10ml,合并二氯甲烷液,蒸干,残渣加二氯甲烷lml使溶解,作为供试品溶液。另取荷叶碱对照品,加二氯甲烷制成每lml含0.3mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,分别吸取上述两种供试品溶液6μl、10μl,分别点于同一硅胶g薄层板上,以正丁醇-醋酸-水(4:1:1)为展开剂,展开,取出,晾干,喷以稀碘化铋钾试液,供试品色谱中,在与对照品色谱相应的位置上,显相同顏色的斑点。
2)补骨脂的鉴别:取本品薄膜衣片2g,研细,加乙醇10ml,超声15分钟,放冷,滤过,滤液蒸干,残渣加乙醇2ml使溶解,作为供试品溶液。取补骨脂对照药材粉末0.5g,同法制得对照药材溶液。另取补骨脂素、异补骨脂素对照品,加乙醇制成每1ml含2mg的混合溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述三种溶液各2~5μl,分别点于同一硅胶g薄层板上,以正己烷-乙酸乙酯(4:1)为展开剂,展开,取出,晾干,喷以10%氢氧化钾乙醇溶液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同的两个荧光斑点。
3)丹参的鉴别:取本品薄膜衣片2g,研细,加乙醇10ml,超声15分钟,过滤,滤液挥干,残渣加乙酸乙酯1ml使溶解,作为供试品溶液。取丹参对照药材粉末0.5g,同法制得对照药材溶液。另取丹参酮ⅱa对照品,加乙醇制成每1ml含1mg的溶液,作为对照品溶液,照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液10μl、对照药材溶液和对照品溶液各5μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶g薄层板上,以石油醚(60~90℃)∶乙酸乙酯(4∶1)为展开剂,展开,取出,晾干,在紫外光灯(365nm)下检视。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色荧光斑点。
4)山楂的鉴别:取本品薄膜衣片1.2g,研细,加甲醇50ml,加热回流30分钟,过滤,滤液挥干,残渣加水20ml使溶解,用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取山楂对照药材4g,加水100ml,提取1小时,滤过,滤液浓缩至20ml,用稀盐酸调节ph值至1~2,用乙酸乙酯振摇提取2次,同法制成对照药材溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取供试品溶液10μl、对照药材溶液20μl,分别点于同一硅胶g薄层板上,以环已烷∶乙酸乙酯∶甲酸(20∶20∶1)为展开剂,展开,取出,晾干,喷以2%三氯化铁乙醇溶液,在105℃加热至斑点显色清晰,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点。
5)番泻叶的鉴别:取本品薄膜衣片1.5g,研细,加甲醇-乙酸乙酯(1:1)的混合溶液15ml,加热回流15分钟,放冷,过滤,滤液蒸干,残渣加甲醇2ml使溶解,作为供试品溶液。取番泻叶对照药材0.5g,加甲醇15ml,加热回流15分钟,放冷,滤过,滤液蒸干,残渣加水15ml使溶解,滤过,滤液用二氯甲烷振摇提取2次,每次15ml,合并二氯甲烷液,用5%碳酸钠溶液25ml振摇提取,弃去二氯甲烷液,碳酸钠溶液用盐酸调节ph值至2,再用二氯甲烷30ml振摇提取,分取二氯甲烷液,用水洗涤2次,每次30ml,二氯甲烷液蒸干,残渣加甲醇1ml使溶解,作为对照药材溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液与对照药材溶液各10μl,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶g薄层板上,以正己烷:乙酸乙酯:甲酸(3∶1∶0.1)为展开剂,展开,取出,晾干。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,置氨蒸气中熏后,斑点变成红色。
【限量测定】
大黄酸:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%甲酸水溶液(31:69)为流动相;检测波长为254nm。理论板数按大黄酸峰计算应不低于5000。
对照品溶液的制备取大黄酸对照品适量,精密称定,加甲醇制成每1ml含1μg的溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声处理20分钟,取出,放至室温,并定容至刻度,摇匀,14000rpm离心10分钟,取上清液,即得。
测定法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
本品每片含番泻叶以大黄酸(c15h18o6)计,不得过0.10mg。
【含量测定】
1)补骨脂素、异补骨脂素和补骨脂苷、异补骨脂苷:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(20:80)为流动相;检测波长为246nm,柱温60℃,流速0.3ml/min。理论板数按补骨脂素、异补骨脂素峰计算应不低于5000。
对照品溶液的制备取补骨脂素、异补骨脂素对照品适量,精密称定,分别加甲醇制成每1ml各含5μg的混合对照品溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声处理15分钟,取出,放至室温,并定容至刻度,摇匀,14000rpm离心10分钟,取上清液,即得。
测定法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
补骨脂苷、异补骨脂苷
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.1%甲酸水溶液(15.7:84.3)为流动相;检测波长为246nm,柱温50℃,流速0.3ml/min。理论板数按补骨脂苷计算应不低于5000。
对照品溶液的制备取补骨脂苷、异补骨脂苷对照品适量,精密称定,加50%甲醇制成每1ml分别含12μg、8μg的混合对照品溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于25ml容量瓶中,加甲醇适量,超声处理20分钟,取出,放至室温,并定容至刻度,摇匀,取5ml置10ml容量瓶中,用10%甲醇定容至刻度,摇匀,14000rpm离心10分钟,取上清液,即得。
测定法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
本品每片含盐补骨脂以游离型补骨脂素(c11h6o3)和异补骨脂素(c11h6o3)总量计,薄膜衣片不得少于0.21mg;本品每片含盐补骨脂以游离型和结合型补骨脂素(c11h6o3)和异补骨脂素(c11h6o3)(补骨脂苷、异补骨脂苷与补骨脂素、异补骨脂素的折算系数0.5082)总量计,薄膜衣片不得多于3.38mg。
2)丹酚酸b:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-0.1%甲酸水溶液(18:82)为流动相;检测波长为286nm,柱温50℃,流速0.3ml/min。理论板数按丹酚酸b峰计算应不低于5000。
对照品溶液的制备取丹酚酸b对照品适量,精密称定,加75%甲醇制成每1ml含29μg的溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.5g,精密称定,置于50ml容量瓶中,加75%甲醇适量,超声处理20分钟,取出,放置室温,并定容至刻度,摇匀,取5ml置10ml容量瓶中50%甲醇定容至刻度,混匀,14000rpm离心10分钟,取上清液,即得。
测定法分别精密吸取上述对照品溶液与供试品溶液各2μl,注入超高效液相色谱仪,测定,即得。
本品每片含丹参以丹酚酸b(c36h30o16)计,薄膜衣片不得少于1.37mg。
3)荷叶碱:
色谱条件与系统适应性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈-水-三乙胺-冰醋酸(27:70.6:1.6:0.78)为流动相;检测波长为270nm。理论板数按荷叶碱峰计算应不低于2000。
对照品溶液的制备取荷叶碱对照品适量,精密称定,加甲醇制成每1ml含16μg的溶液,即得。
供试品溶液的制备取本品薄膜衣片24片,研细,取约0.4g,精密称定,用加浓氨试液5ml湿润,加三氯甲烷60ml,加热回流3小时,放冷,移至分液漏斗中,分取三氯甲烷层,余留物再用三氯甲烷25ml分3次振摇提取,合并三氯甲烷液,回收三氯甲烷至干,残渣用乙醇溶解,转移至10ml量瓶中,加乙醇至刻度,滤过,取续滤液,即得。
测定法分别精密吸取对照品溶液与供试品溶液各20μl,注入液相色谱仪,测定,即得。
本品每片含荷叶以荷叶碱(c19h21no3)计,薄膜衣片不得少于0.25mg。
- 还没有人留言评论。精彩留言会获得点赞!