一种依巴斯汀中有关物质的检测方法与流程
本发明涉及药物化学分析领域,具体涉及一种依巴斯汀中有关物质的检测方法。
背景技术:
依巴斯汀(ebastine),化学名为1-[4-(1,1-二甲基乙基)苯基]-4-[4-(二苯基甲氧基)-1-哌啶基]-1-丁酮,其分子式为c32h39no2,分子量为469.67,结构式如下所示:
依巴斯汀又名开思亭;艾巴停;司可停,通常状态下为非吸湿性、白色结晶粉末。依巴斯汀是组胺h1受体阻断剂,其抗组胺活性在低浓度时有竞争性,高浓度时是非竞争性的。临床上用于变应性疾病,包括儿童变应性鼻炎、成人的终年鼻炎、季节性鼻炎、枯草热和慢性荨麻疹等。
依巴斯汀对h1受体有高度的选择性,无中枢抑制作用。与特非那定相比,依巴斯汀作用强而持久。依巴斯汀对组胺诱发的支气管痉挛具有保护作用,动物实验证实其保护作用是特非那定的4.5倍,阿斯米唑的2.9倍,连续用药不蓄积。其代谢产物carebestine对组胺诱发的支气管痉挛的保护作用是原药的3倍。依巴斯汀具有拮抗白三烯c4的作用,可抑制白三烯c4诱发的支气管痉挛,有抗胆碱作用。可抑制试验性喘息和鼻过敏。。
在依巴斯汀的原料药生产或制剂过程当中,会产生一些杂质,沿用了欧洲药典的编号,目前所知的主要杂质如下表所示:
依巴斯汀欧洲药典9.6版中列举了杂质a~杂质h共8个杂质,但根据依巴斯汀的工艺(如下式所示),上述杂质并不是依巴斯汀有关物质研究的所有杂质。对依巴斯汀工艺进行分析:原料对叔丁基-4-氯苯丁酮可能残留;原料对叔丁基-4-氯苯丁酮易发生副反应生成杂质3a和杂质3b;原料对叔丁基-4-氯苯丁酮可能残留有结构类似物4-氯苯丁酮,最后生成依巴斯汀结构类似物杂质i;中间体4-(二苯甲氧基)哌啶可能残留有过度反应产物杂质j。经过上述分析,杂质i、杂质j、杂质3a、杂质3b、对叔丁基-4-氯苯丁酮在依巴斯汀有关物质研究中必不可少。现有的分析技术无法对依巴斯汀及13中杂质进行有效检测分离。
在欧洲药典9.0,韩国药典第十版中公开了一种检测方法,其采用乙腈-磷酸-氢氧化钠溶液进行等度洗脱,其ph为5.0,其使用的色谱柱是氰基硅胶柱(4.6×250mm,5μm)。且其分析时间周期长达3个小时,主峰约110分钟出峰,且拖尾严重,测定很不方便,不利于实际推广应用。
我国的国家药品标准方法,采用甲醇-醋酸铵缓冲液(ph3.9)为流动相,检测波长为254nm,但该波长下个别杂质无吸收,且杂质无法达到有效分离,不能准确检测杂质。
在欧洲药典9.6,英国药典2019版上也公开了一种依巴斯汀原料药的检测方法,其描述的方法采用等度洗脱的方式,存在部分杂质无法有效分离的情况。
因此,在现今技术存在上述问题的情况下,需要对依巴斯汀有关物质分析方法进行改进,建立一个快速、有效的检测分离方法,以区分并检测分离出依巴斯汀原料药内的有关物质。
技术实现要素:
本申请的主要目的是提供一种依巴斯汀中有关物质的检测方法。
我方技术方案是基于在现有技术中存在着不能有效检测分离依巴斯汀原料药中各杂质的问题进行的改进,并摸索优化了相关方案而成的。
本申请采用了以下技术方案:
一种依巴斯汀中有关物质的检测方法,所使用的色谱柱为c18色谱柱,流动相分为a和b两相,流动相a由ph值为5.7的磷酸缓冲溶液和甲醇组成,流动相b由乙腈和甲醇所组成,并进行梯度洗脱。
优选的,ph值为5.7。
优选的,流动相a中磷酸缓冲溶液与甲醇的体积比为25:75,流动相b中乙腈与甲醇的体积比为42:38。
优选的,梯度洗脱的程序如下表所示:
本申请的另外一种较为优选的方案如下:
一种依巴斯汀中有关物质的检测方法,包括以下步骤:
(1)供试品溶液的制备:取依巴斯汀原料适量,用溶剂稀释成每1ml中约含2.0~3.0mg的溶液,作为供试品溶液;
(2)对照溶液的制备:量取供试品溶液,用稀释液定量稀释制成每1ml含2.0~3.0μg的溶液,作为对照溶液;
(3)测定:采用c18反相色谱柱,以加入二乙胺调节ph值为5.7的磷酸缓冲溶液和甲醇为流动相a,磷酸缓冲溶液和甲醇的体积比为25:75,以乙腈和甲醇为流动相b,乙腈和甲醇的体积比为42:38,使用梯度洗脱,梯度洗脱的程序如下所示:
取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图;
进一步的,供试品溶液的浓度为2.5mg/ml。
进一步的,其中液相色谱的分析条件为以下(ⅰ)至(ⅵ)中的一项或者多项:
(ⅰ)色谱柱的规格为4.6*150mm,5μm,
(ⅱ)色谱柱的型号为shim-packclc-ods(m)c18,
(ⅲ)色谱柱温度为40±5℃,
(ⅳ)流动相比例的变换范围:±2%,
(ⅴ)流速为0.8~1.2ml/min,
(ⅵ)进样量为10~30μl,
(ⅶ)检测波长为210nm或252nm。
本申请还包括了此检测方法在药物杂质检测分离当中的应用。
由于采用以上技术方案,本申请的有益效果在于:
1.在我方申请的条件下,依巴斯汀原料药中的有关物质比现有技术多,对于原料的分析更加全面且均能有效检测分离,而且检测分析的周期较之前短,节约了检测的时间。
2.操作简便,适于实际应用和推广。
附图说明
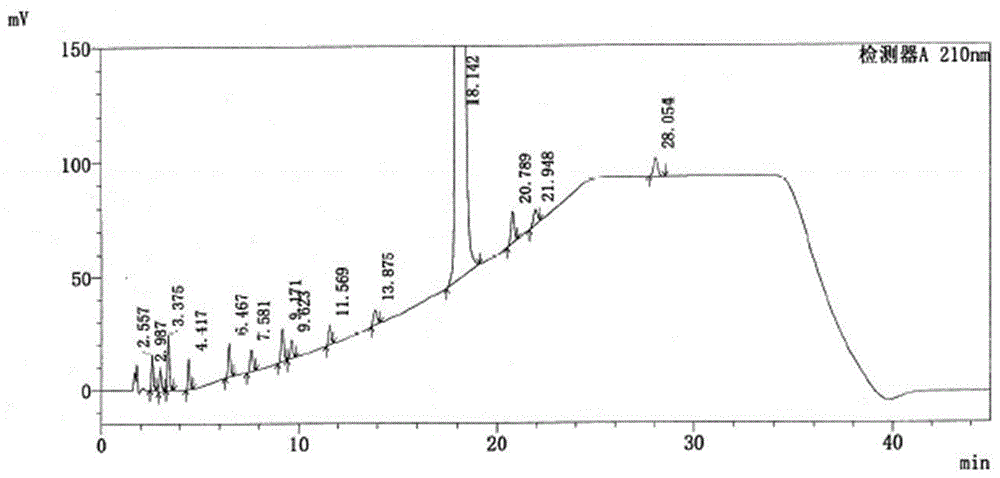
图1是我方实施例1的依巴斯汀样品在210nm波长下的检测色谱图;
图2是我方实施例1的依巴斯汀样品在252nm波长下的检测色谱图;
图3是我方实施例2的依巴斯汀样品在流速为0.8ml/min在210nm波长下的检测色谱图;
图4是我方实施例2的依巴斯汀样品在流速为0.8ml/min在252nm波长下的检测色谱图;
图5是我方实施例2的依巴斯汀样品在流速为1.2ml/min在210nm波长下的检测色谱图;
图6是我方实施例2的依巴斯汀样品在流速为1.2ml/min在252nm波长下的检测色谱图;
图7是我方实施例3的依巴斯汀样品在柱温为35℃下在210nm波长下的检测色谱图;
图8是我方实施例3的依巴斯汀样品在柱温为35℃下在252nm波长下的检测色谱图;
图9是我方实施例3的依巴斯汀样品在柱温为45℃下在210nm波长下的检测色谱图;
图10是我方实施例3的依巴斯汀样品在柱温为45℃下在252nm波长下的检测色谱图;
图11是对比实施例1的色谱条件下依巴斯汀样品的检测色谱图;
图12是对比实施例2的色谱条件下依巴斯汀样品的检测色谱图。
具体实施方式
下面通过具体实施例和附图对本申请作进一步详细说明。以下实施例仅对本申请进行进一步说明,不应理解为对本申请的限制。
实施例1
一种依巴斯汀中有关物质的检测方法,包括以下步骤:
(1)供试品溶液的制备:取依巴斯汀原料适量,用溶剂稀释成每1ml中约含2.5mg的溶液,作为供试品溶液;
(2)对照溶液的制备:量取供试品溶液,用稀释液定量稀释制成每1ml含2.5μg的溶液,作为对照溶液;
(3)流动相的制备:量取800ml水,加磷酸12ml,二乙胺12ml,用二乙胺调节ph至5.7后,用水稀释至1000ml,制备得到流动相a,按照体积比42:38量取乙腈溶液和甲醇溶液,配置成流动相b;
(4)测定:采用c18反相色谱柱,以加入二乙胺调节ph值为5.7的磷酸缓冲溶液和甲醇为流动相a,磷酸缓冲溶液和甲醇的体积比为25:75,以乙腈和甲醇为流动相b,乙腈和甲醇的体积比为42:38,使用梯度洗脱,梯度洗脱的程序如下所示:
取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图;
所要检测分离的有关物质如下表所示:
其中,液相色谱的条件如下:
色谱柱:shim-packclc-ods(m)c18(4.6*150mm*5μm),色谱柱温度为40℃,流速为1.0ml/min,进样量为10μl,检测波长为210nm以及252nm。
经检测,色谱图如图1和图2所示,图1为在210nm波长下的检测图谱,图2为在252nm波长下的检测图谱,从图1和图2中可以看到,依巴斯汀原料药中的13种杂质和依巴斯汀均完全检测分离,峰形齐整不拖尾,检测时间45min,证明本申请提供的方法,检测效果好、速度快,完全符合实际需要。此实施例为最佳实施例,在同样的条件下,实验6次,实验的重复性良好。
实施例2
一种依巴斯汀中有关物质的检测方法,包括以下步骤:
(1)供试品溶液的制备:取依巴斯汀原料适量,用溶剂稀释成每1ml中约含2.5mg的溶液,作为供试品溶液;
(2)对照溶液的制备:量取供试品溶液,用稀释液定量稀释制成每1ml含2.5μg的溶液,作为对照溶液;
(3)流动相的制备:量取800ml水,加磷酸12ml,二乙胺12ml,用二乙胺调节ph至5.7后,用水稀释至1000ml,制备得到流动相a,按照体积比42:38量取乙腈溶液和甲醇溶液,配置成流动相b;
(4)测定:采用c18反相色谱柱,以加入二乙胺调节ph值为5.7的磷酸缓冲溶液和甲醇为流动相a,磷酸缓冲溶液和甲醇的体积比为25:75,以乙腈和甲醇为流动相b,乙腈和甲醇的体积比为42:38,使用梯度洗脱,梯度洗脱的程序如下所示:
取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图;
所要检测分离的有关物质同实施例1表中所示。
其中,液相色谱的条件如下:
色谱柱:shim-packclc-ods(m)c18(4.6*150mm*5μm),色谱柱温度为40℃,流速分别设为0.8ml/min及1.2ml/min,进样量为10μl,检测波长为210nm以及252nm。
经检测,色谱图如图3~图6所示,图3为流速为0.8ml/min在210nm波长下的检测图谱,图4为流速为0.8ml/min在252nm波长下的检测图谱,图5为流速为1.2ml/min在210nm波长下的检测图谱,图6为流速为1.2ml/min在252nm波长下的检测图谱。由图3~图6可以看到,依巴斯汀原料药中的13种杂质和依巴斯汀均完全检测分离,峰形齐整不拖尾,检测时间45min,证明本申请提供的方法,检测效果好、速度快,完全符合实际需要。
实施例3
一种依巴斯汀中有关物质的检测方法,包括以下步骤:
(1)供试品溶液的制备:取依巴斯汀原料适量,用溶剂稀释成每1ml中约含2.5mg的溶液,作为供试品溶液;
(2)对照溶液的制备:量取供试品溶液,用稀释液定量稀释制成每1ml含2.5μg的溶液,作为对照溶液;
(3)流动相的制备:量取800ml水,加磷酸12ml,二乙胺12ml,用二乙胺调节ph至5.7后,用水稀释至1000ml,制备得到流动相a,按照体积比42:38量取乙腈溶液和甲醇溶液,配置成流动相b;
(4)测定:采用c18反相色谱柱,以加入二乙胺调节ph值为5.7的磷酸缓冲溶液和甲醇为流动相a,磷酸缓冲溶液和甲醇的体积比为25:75,以乙腈和甲醇为流动相b,乙腈和甲醇的体积比为42:38,使用梯度洗脱,梯度洗脱的程序如下所示:
取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图;
所要检测分离的有关物质同实施例1表中所示。
其中,液相色谱的条件如下:
色谱柱:shim-packclc-ods(m)c18(4.6*150mm*5μm),色谱柱温度分别设为35℃及45℃,流速为1.0ml/min,进样量为10μl,检测波长为210nm以及252nm。
经检测,色谱图如图7~图10所示,图7为柱温在35℃在210nm波长下的检测图谱,图8为柱温在35℃在254nm波长下的检测图谱,图9为柱温在45℃在210nm波长下的检测图谱,图10为柱温在45℃在254nm波长下的检测图谱。由图7~图10可以看到,依巴斯汀原料药中的13种杂质和依巴斯汀均完全检测分离,峰形齐整不拖尾,检测时间45min,证明本申请提供的方法,检测效果好、速度快,完全符合实际需要。
对比实施例1
按照依巴斯汀有关物质测定——欧洲药典(ep9.0版)方法测定:
供试品溶液配制:称取供试品约125.0mg,精密称定,置50ml量瓶中,加混合溶液[乙腈∶(1.1g/l磷酸用40g/l氢氧化钠溶液调节ph值5.0)=65∶35]溶解并稀释至刻度,摇匀,过滤,即得。(2.5mg/ml)
其中,液相色谱的条件如下:
色谱柱:agilentsb-cn(4.6*250mm*5μm),流动相:乙腈∶(1.1g/l磷酸用40g/l氢氧化钠溶液调节ph值5.0)(35∶65),流速为1.0ml/min,进样量为10μl,检测波长为210nm。
经检测,色谱图如图11所示,由色谱图可知依巴斯汀的保留时间是106min,根据色谱收集时间要求(依巴斯汀保留时间的1.4倍),整个收集时间需要170min,约3个小时;依巴斯汀主峰严重拖尾,峰比较宽(45~50min),不符合实际需要。
对比实施例2
按照依巴斯汀有关物质测定——欧洲论坛28.3方法测定:
供试品溶液配制:称取供试品约125mg及各杂质适量,精密称定,置50ml量瓶中,加5ml甲醇溶解,用混合溶液[甲醇∶(磷酸缓冲溶液(取800ml水,加12ml的磷酸,12ml二乙胺,用二乙胺调ph6.0,加水稀释至1000ml)=50∶50]稀释至刻度,摇匀,即得。
其中,液相色谱的条件如下:
色谱柱:inertsilods-3(4.6*250mm*5μm),流动相a-乙腈-甲醇(20∶42∶38);流动相a:磷酸缓冲溶液(取800ml水,加12ml的磷酸,12ml二乙胺,用二乙胺调ph6.0,加水稀释至1000ml)。流速:1.0ml/min;检测波长:210nm;柱温:40℃。
经检测,色谱图如图12所示,由色谱图可知,色谱图中只有12个峰,而溶液中有14个成分,说明部分杂质重合,不符合实际需要。
以上内容是结合具体的实施方式对本申请所作的进一步详细说明,不能认定本申请的具体实施只局限于这些说明。对于本申请所属技术领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!