一种同时分离、定量多种组分的方法与流程
本发明属于分析技术领域,主要涉及一种同时分离、定量多种组分的方法,更具体地,涉及一种对多组分中的丙烯酸树脂、聚维酮和聚乙二醇同时分离、定量的方法。
背景技术:
丙烯酸树脂(polymethacrylates),商品名为尤特奇(eudragit),它包括甲基丙烯酸共聚物和甲基丙烯酸酯共聚物。尤特奇广泛用于药物制剂的胃溶包衣、肠溶包衣、缓控释包衣、保护隔离包衣、缓释骨架材料和经皮给药制剂的骨架胶粘材料。自从第一个产品问世至今已有半个多世纪,已广泛用于各种药物制剂,成为许多国际名牌制剂产品的重要辅料。目前已报道丙烯酸树脂检测方法主要有滴定法[methacrylicacidandethylacrylatecopolymer[s].usp42-nf37,2018:5823]和凝胶色谱法[周五端,赵立那,曹沙沙.丙烯酸树脂分子量及其分布的测定[j].广州化工,2014,42(24):103-104]。
聚维酮(povidone)在医药上有广泛的应用,主要应用于固体制剂中,应用最广的是片剂、颗粒剂的粘合剂。聚维酮也可用作崩解剂、增溶剂、包衣材料,以及助悬剂、稳定剂或增粘剂等。目前已报道聚维酮检测方法主要有氮测定法[聚维酮k30[s].中国药典2015版四部,2015:631等]、采用反相硅胶c18作为固定相的高效液相色谱法[龙佳坤,江洁冰,赵美艳,等.hplc法测定聚维酮碘口服液中聚维酮的含量[j].中国临床药学杂志,2016,25(5):300-302等]
聚乙二醇(polyethyleneglycol,peg)在医药工业中作为赋形剂,用作栓剂、膏剂的制备。聚乙二醇也可用作助悬剂、增塑剂、润滑剂等。适用于医药、化肥、造纸、陶瓷、洗涤剂、化妆品、热处理、水处理、消防、石油开采等多种行业。目前已报道聚乙二醇检测方法主要有凝胶色谱法[polyethyleneglycol3350[s].usp42-nf37,2017:3557等]、疏水作用色谱法[jameservinseely,louisville,colo.useofhydrophobicinteractionchromatographytopurifypolyethyleneglycols(p).us005747639a,1998-05-05]、采用c18、c8、c4反相硅胶作为固定相的高效液相色谱法[聚乙氧基化非离子表面活性剂中聚乙二醇含量的测定高效液相色谱法(征求意见稿)[j].日用化学品科学,2015,38(6):50-52等]、分光光度法[聚乙二醇残留量测定法[s].中国药典2015版四部,2015:238等]。
目前暂无文献报道方法可对丙烯酸树脂、聚维酮、聚乙二醇等多种组分同时分离、定量检测。已报道的丙烯酸树脂、聚维酮、聚乙二醇的检测方法(凝胶渗透色谱法、滴定法、氮测定法、反相硅胶或疏水作用色谱法、分光光度法等)专属性较差,其他组分易对测定产生干扰,不能多种组分同时分离、定量。①测定丙烯酸树脂含量:采用酸碱滴定原理,很多酸性组分易对方法测定产生干扰;采用已报道的液相凝胶色谱法,聚乙二醇等组分易对丙烯酸树脂测定产生干扰,且不能实现多种组分同时分离定量。②测定聚维酮含量:采用氮测定法进行聚维酮的含量检测时,样品中很多组分均含有氮元素,易产生干扰;采用文献报道的反相硅胶c18作为固定相的高效液相色谱法,采用紫外检测器进行检测时,仅可实现聚维酮的含量测定,无法实现多种组分的同时分离、定量检测。③测定聚乙二醇含量:采用已报道的液相凝胶色谱法,聚维酮等组分易与聚乙二醇色谱峰重叠,无法分离和定量;采用疏水作用色谱法或采用反相硅胶c18、c8、c4作为固定相的高效液相色谱法,因样品中很多组分为强极性化合物,均在溶剂峰前后出峰,很难实现多种组分的同时分离;采用分光光度法,操作麻烦(需要衍生化),易受其他组分干扰,专属性差且线性范围窄,无法实现多种组分同时分离、定量。
同时,现有凝胶渗透色谱法,在检测丙烯酸树脂等组分的分子量时,流动相若使用n,n-二甲基甲酰胺等强极性试剂为流动相,为消除键合作用往往向强极性试剂中加入溴化锂、氯化锂等盐,溴化锂、氯化锂等盐对色谱柱有较大伤害。
针对上述检测方法专属性较差、不能多种组分同时检测的问题,本发明提供了一种同时分离、定量多种组分的方法。该方法专属性良好,可使丙烯酸树脂、聚维酮、聚乙二醇等多种组分得到较好的分离并可准确的定量分析。为药物制剂以及其他产品(如化妆品、食品)的逆向工程、组分分析、辅料的放行检测提供了良好的方法。同时,本方法无需在流动相中添加溴化锂、氯化锂等盐,显著降低色谱柱的损耗。
技术实现要素:
发明目的:针对现有技术存在的问题,本发明提供了一种同时分离、定量多种组分的方法,特别是一种对多组分中的丙烯酸树脂、聚维酮和聚乙二醇同时分离、定量的方法。
为解决上述技术问题,本发明采取如下技术方案:
一种同时分离、定量多种组分的方法,尤其是一种对多组分中的丙烯酸树脂、聚维酮和聚乙二醇同时分离、定量的方法,包括如下步骤:
(1)将待测样品进行前处理,制备样品溶液;
(2)用凝胶渗透色谱法对步骤(1)得到的样品进行测定,并分析样品溶液的测定结果;其中,色谱柱为usp液相色谱柱分类l21;流动相包括二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、n-甲基吡咯烷酮(nmp)、二甲基乙酰胺(dmac)中的任意一种;检测器为通用型检测器。优选地,样品溶液各组分的定量方法均为外标法进行定量。
其中,所述样品为多组分混合物,所述混合物包括但不限于丙烯酸树脂、聚维酮、聚乙二醇、原料药、柠檬酸三乙酯、微晶纤维素、十二烷基硫酸钠、滑石粉、硬脂酸镁、富马酸、交联聚维酮、糖精钠、无水二氧化硅、硬脂酸镁、氢氧化钠、草莓味香精、甘草味香精、无水二氧化硅中的任意一种或几种的组合。
在一些实施方案中,所述丙烯酸树脂型号包括但不限于尤特奇l100-55、尤特奇l100、尤特奇l30d-55;聚维酮型号包括但不限于聚维酮k30(pvpk30)、聚维酮k90(pvpk90)、聚维酮k29/k32(pvpk29/k32)、聚维酮s630(pvps630);聚乙二醇型号包括但不限于聚乙二醇400(peg400)、聚乙二醇600(peg600)、聚乙二醇6000(peg6000)、聚乙二醇8000(peg8000)。
优选地,组分可以是药用辅料级别,也可以是食品或化妆品用级别。
具体地,所述前处理的步骤包括:将待测样品加至容量瓶中,加入流动相,超声使样品溶解或分散均匀,然后定容过滤即可。
优选地,超声时间为5~30min;定容后溶液通过直径为0.22μm或0.45μm的聚四氟乙烯或等同滤膜过滤。
优选地,所述流动相不含溴化锂(libr)或氯化锂(licl)。
优选地,所述样品经前处理后,丙烯酸树脂浓度为0.02mg/ml~10mg/ml;聚维酮浓度为0.1~10mg/ml;聚乙二醇浓度为0.01~10mg/ml。丙烯酸树脂、聚维酮、聚乙二醇在样品中的具体含量对测定无影响,只需要处理后在上述浓度范围内即可。
步骤(2)中,凝胶渗透色谱法的流速为0.2~2ml/min,进样量为5-100μl,柱温为15~40℃。更优选地,凝胶渗透色谱法的流速为0.6ml/min,进样量为20μl,柱温为35℃。
所述色谱柱为usp液相色谱柱分类l21系列色谱柱中的agilentplgel系列、tosohbiosciencetskgelhxlandhhr系列、phenomenexphenogel系列或waterscorp.styragelhr系列。
在一些实施例中,色谱柱为单根色谱柱;在另一些实施例中,色谱柱为多根色谱柱的串联。更优选地,所述色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根或三根串联。上述色谱柱以刚性苯乙烯-二乙烯基苯共聚物微球(直径3-30μm)为填充剂。
优选地,所述通用型检测器包括示差折光检测器(rid)、蒸发光散射检测器(elsd)。
有益效果:与现有技术相比,本发明具有如下优点:
(1)本发明将至少三种组分(丙烯酸树脂、聚维酮、聚乙二醇)同步进行分离定量,大幅度提高检测效率;
(2)本发明专属性较好,可避免多种组分,尤其是容易相互干扰的组分(如聚乙二醇和聚维酮)对测定的干扰;
(3)本发明操作简单,线性范围宽,定量限低,准确度高;
(4)本方法无需在流动相中添加溴化锂、氯化锂等盐,显著降低色谱柱的损耗。
附图说明
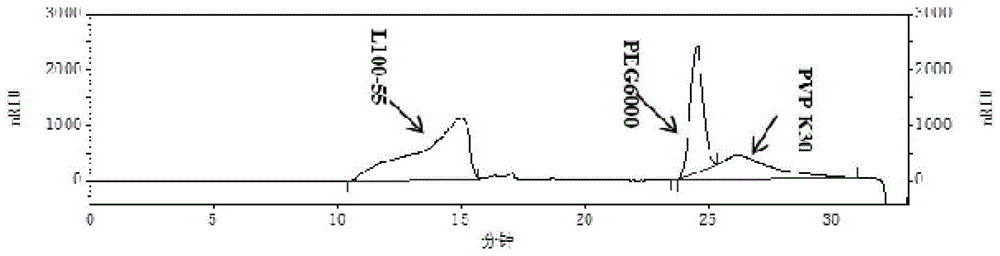
图1为l100-55、peg6000、pvpk30对照品溶液色谱图;
图2为专属性测试中的叠加色谱图;
图3为l100-55线性考察结果;
图4为peg6000线性考察结果;
图5为pvpk30线性考察结果;
图6为流动相dmf中加入不同浓度溴化锂对分离度的影响;
图7为实施例8、实施例9、实施例10中文献色谱条件的色谱结果。
具体实施方式
本发明提出了一种同时对多组分中的丙烯酸树脂、聚维酮、聚乙二醇进行分离、定量测定的方法。
具体地,包括如下步骤:
(1)将含有丙烯酸树脂、聚维酮和聚乙二醇的待测样品溶解于流动相中,流动相包括二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮、二甲基乙酰胺中的任意一种,超声处理使其溶解,然后过滤得到样品溶液;
(2)用凝胶渗透色谱法对步骤(1)得到的样品溶液进行测定,并分析样品溶液的测定结果;其中,色谱柱为usp液相色谱柱分类l21;检测器为通用型检测器。
下面通过具体的实施例详细说明本发明。
实施例1:l100-55、peg6000、pvpk30的同时分离、定量分析。
(1)取l100-55、peg6000、pvpk30各15mg,置同一20ml量瓶中,用dmf溶解并稀释至刻度,摇匀即得对照品溶液。取待测样品(罗红霉素分散片,规格50mg,商品名rulid,根据rulid说明书,该罗红霉素分散片包含辅料微晶纤维素、富马酸、甲基丙烯酸树脂、交联聚维酮、聚乙二醇6000、滑石粉、糖精钠、无水二氧化硅、硬脂酸镁、柠檬酸三乙酯、十二烷基硫酸钠、氢氧化钠、草莓味香精、甘草味香精等)2片,加dmf至容量瓶的2/3体积,超声15min使样品分散均匀(保证样品中l100-55、peg6000、pvpk30可全部溶解),加dmf定容至刻度,摇匀过滤即得样品溶液。
(2)用凝胶渗透色谱法对步骤(1)得到的对照品溶液和样品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根串联;流动相为dmf,检测器为示差折光检测器(rid),柱温和检测器温度为35℃,流速为0.6ml/min,样品进样量为20μl。
对照品溶液色谱图如图1所示,l100-55与peg6000、peg6000与pvpk30分离度分别为4.6、0.7,可实现l100-55、peg6000、pvpk30三组分的同时分离、定量分析。
样品溶液检测结果显示,罗红霉素分散片rulid中含l100-5511.9mg/片、含peg60006.7mg/片,罗红霉素分散片中未检出pvpk30。
实施例2:检测方法的专属性、线性、准确度、定量限的研究。
(一)专属性:
分别对流动相(dmf)、专属性溶液(所述专属性溶液含原料药(罗红霉素)、微晶纤维素、富马酸、交联聚维酮、滑石粉、糖精钠、无水二氧化硅、硬脂酸镁、柠檬酸三乙酯、十二烷基硫酸钠、氢氧化钠、草莓味香精、甘草味香精)进样检测,实验结果如图2所示,结果表明流动相、专属性溶液对l100-55、peg6000、pvpk30的测定均无干扰。
(二)线性:
配制系列线性溶液进样,以浓度为横坐标(x)、峰面积为纵坐标(y),进行线性回归,得线性回归方程(如图3-图5所示),l100-55、peg6000、pvpk30相关系数的平方(r2)分别为1.000、1.000、0.999(均不小于0.99)。即l100-55、peg6000、pvpk30浓度在0.1mg/ml~10mg/ml范围内,均显示了可接受的线性。
(三)准确度:
制备含peg6000、pvpk30、l100-55不同含量的片剂,根据药用辅料手册(r.c.罗、p.j,舍斯基、p.j.韦勒编,刘俊民主译):聚乙二醇在药物制剂的浓度为0~30%;聚维酮在药物制剂的浓度为0~25%;丙烯酸树脂在药物制剂的浓度为0~50%。片剂中除peg6000、pvpk30、l100-55外,包含常规量的原料药(罗红霉素)、微晶纤维素、富马酸、交联聚维酮、滑石粉、糖精钠、无水二氧化硅、柠檬酸三乙酯、十二烷基硫酸钠,改变片剂中peg6000、pvpk30、l100-55的加入含量,制备样品溶液,使用该方法确认方法的回收率。结果显示回收率在86-108%之间,该方法准确度良好。数据结果如表1所示。
表1l100-55、peg6000、pvpk30准确度结果
(四)定量限的测定:
配制不同浓度的样品溶液,记录当信噪比约为10的浓度为定量限(loq)浓度,重复进样6针loq溶液色谱图中主峰的峰面积rsd应不大于10%。数据结果如表2所示。
表2l100-55、peg6000、pvpk30定量限、检测限结果
实验得到:l100-55的定量限为25.1μg/ml;peg6000的定量限为12.5μg/ml;pvpk30的定量限为100.9μg/ml。
实施例3:流动相dmf中加入不同浓度溴化锂对分离度的影响
本实施例除了将实施例1中的流动相和稀释剂由dmf换为dmf(含0.01%libr)、dmf(含0.05%libr)、dmf(含0.1%libr)外,其他方法步骤均同实施例1进行对照品溶液检测。
色谱图如图6所示,分离度数据见表3,若流动相中加入溴化锂等盐,l100-55、peg6000、pvpk30三者间分离度变差,不可完全分离,不能对l100-55、peg6000、pvpk30三种组分同时分离、定量。
表3流动相dmf中加入不同浓度溴化锂对分离度的影响
实施例4:不同型号丙烯酸树脂、聚乙二醇、聚维酮的检测
(1)取l100-55、peg400、pvpk29/k32各15mg,置同一20ml量瓶中,用dmf溶解并稀释至刻度,摇匀即得对照品溶液1;取l100、peg600、pvpk90各15mg,置同一20ml量瓶中,用dmf溶解并稀释至刻度.摇匀即得对照品溶液2;取l30d-55、peg8000、pvps630各15mg,置同一20ml量瓶中,用dmf溶解并稀释至刻度.摇匀即得对照品溶液3;
(2)用凝胶渗透色谱法对步骤(1)得到的对照品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根串联;流动相为dmf,检测器为示差折光检测器(rid),柱温为35℃,流速为0.6ml/min,样品进样量为20μl。
进样结果显示,对照品溶液1中,l100-55与pvpk29/k32、pvpk29/k32与peg400分离度分别为3.8、1.9;对照品溶液2中,l100与pvpk90、pvpk90与peg600分离度分别为4.2、1.7;对照品溶液3中,l30d-55与peg8000、peg8000与pvps630分离度分别为3.7、0.6。说明该分析方法可实现丙烯酸树脂、聚乙二醇、聚维酮三组分的同时定量分析。
实施例5:使用不同流动相进行多组分检测
(1)取l100-55、peg6000、pvpk30各15mg,置同一20ml量瓶中,平行制备3份,分别用二甲基亚砜(dmso)、n-甲基吡咯烷酮(nmp)、二甲基乙酰胺(dmac)溶解并稀释至刻度,摇匀即得相应对照品溶液。
(2)用凝胶渗透色谱法对步骤(1)得到的对照品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根串联;流动相分别为二甲基亚砜(dmso)、n-甲基吡咯烷酮(nmp)、二甲基乙酰胺(dmac),检测器为蒸发光散射检测器(elsd),柱温为35℃,流速为0.6ml/min,样品进样量为20μl。
进样结果显示,当流动相分别为二甲基亚砜(dmso)、n-甲基吡咯烷酮(nmp)、二甲基乙酰胺(dmac)时,可实现l100-55、peg6000、pvpk30三组分的同时定量分析。
实施例6:使用不同检测器进行多组分检测
(1)取l100-55、peg6000、pvpk30各15mg,置同一20ml量瓶中,用dmf溶解并稀释至刻度,摇匀即得对照品溶液。
(2)用凝胶渗透色谱法对步骤(1)得到的对照品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根串联;流动相为dmf,检测器为蒸发光散射检测器(elsd),柱温为35℃,流速为0.6ml/min,样品进样量为20μl。
进样结果显示,当检测器为elsd时,l100-55与peg6000、peg6000与pvpk30分离度分别为4.0、0.7,可实现l100-55、peg6000、pvpk30三组分的同时定量分析。
实施例7:
(1)取l100-55、peg6000、pvpk30适量置同一量瓶中,制备成如表4所示各浓度样品溶液。
(2)用凝胶渗透色谱法对步骤(1)得到的样品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根串联;流动相为dmf,检测器为示差折光检测器(rid),柱温为35℃,流速为0.6ml/min,样品进样量为20μl。
进样结果如表4显示,l100-55与peg6000的分离度均大于3.7、peg6000与pvpk30分离度均大于0.5,三组分的回收率均在93~103%之间,可实现l100-55、peg6000、pvpk30三组分的同时定量分析。
表4不同浓度l100-55、peg6000、pvpk30检测结果
实施例8:
参照文献1[周五端,赵立那,曹沙沙.丙烯酸树脂分子量及其分布的测定[j].广州化工,2014,42(24):103-104]。
(1)取l100-55、peg6000、pvpk30各15mg,分别置不同20ml量瓶中,用四氢呋喃(thf)溶解并稀释至刻度,摇匀,即得各组分对照品溶液。取peg6000、l100-55、pvpk30各15mg,置同一20ml量瓶中,用四氢呋喃(thf)溶解并稀释至刻度,摇匀,即得混合对照品溶液。
(2)用凝胶渗透色谱法对步骤(1)得到的对照品溶液和混合对照品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为安捷伦plgel5μmmixed-d,7.5×300mm,两根串联;流动相为thf,检测器为示差折光检测器(rid),柱温和检测器温度为32℃,流速为0.3ml/min,样品进样量为20μl。
实验发现:①pvpk30不可溶于thf,故pvpk30无峰流出;②peg6000在thf中溶解性较差,极微溶于thf(<0.4mg/ml),不利于peg6000的定量分析。
色谱图如图7文献1所示,pvpk30未检出,peg6000峰面积较本专利方法明显变小,l100-55与peg6000间分离度变差(本专利方法,分离度为4.6;文献1,分离度为1.2),此条件不能实现对l100-55、peg6000、pvpk30三种组分同时分离、定量检测。
实施例9:
参照文献2[龙佳坤,江洁冰,赵美艳等.hplc法测定聚维酮碘口服液中聚维酮的含量[j].中国临床药学杂志,2016,25(5):300-302]。
(1)取pvpk30对照品约0.01g,精密称定,置10ml容量瓶中,加水溶解,定容至刻度,摇匀,即得系统适用性溶液;取peg6000、l100-55、pvpk30各15mg,分别置不同20ml量瓶中,加水溶解,定容至刻度,摇匀,即得各组分对照品溶液;取peg6000、l100-55、pvpk30各15mg,置同一20ml量瓶中,加水溶解,定容至刻度,摇匀,即得混合对照品溶液。
(2)用高效液相色谱法对步骤(1)得到的系统适用性溶液、各组分对照品溶液、混合对照品溶液进行测定,并分析测定结果;
其中,高效液相色谱条件为:
色谱柱为phenomenexc18,150×4.6mm,5μm;流动相为乙腈:水=5:95;检测器为紫外检测器,检测波长为205nm;柱温为20℃;流速为0.8ml/min;样品进样量为20μl。
色谱图如图7文献2所示,在该色谱条件下,系统适用性通过,l100-55、peg6000均无明显出峰,仅可对pvpk30进行定量检测。故,此条件不能实现对l100-55、peg6000、pvpk30三种组分同时定量检测。
实施例10:
参照文献3[polyethyleneglycol3350[s].usp42-nf37,2017:3557]。
(1)取peg6000对照品约0.4g,精密称定,置20ml容量瓶中,用流动相(50μg/mlnan3水溶液)溶解,定容至刻度,摇匀,即得系统适用性溶液;取l100-55、peg6000、pvpk30各15mg,分别置不同20ml量瓶中,用流动相(50μg/mlnan3水溶液)溶解并稀释至刻度,摇匀,即得各组分对照品溶液。取peg6000、l100-55、pvpk30各15mg,置同一20ml量瓶中,用流动相(50μg/mlnan3水溶液)溶解并稀释至刻度,摇匀,即得混合对照品溶液。
(2)用凝胶渗透色谱法对步骤(1)得到的系统适用性溶液、各组分对照品溶液、混合对照品溶液进行测定,并分析测定结果;
其中,色谱条件为:
色谱柱为tskgelg2500pwxl,7.8×300mm,7μm;流动相为50μg/mlnan3水溶液;检测器为示差折光检测器;柱温和检测器温度为35℃;流速为0.8ml/min;样品进样量为20μl。
色谱图如图7文献3所示,在该色谱条件下,系统适用性通过,l100-55无峰流出,peg6000和pvpk30色谱峰完全重叠,无法分离和定量。
实施例11:
参照文献4[聚乙二醇残留量测定法[s].中国药典2015版四部,2015:238]。
(1)取l100-55、peg6000、pvpk30各5mg,分别置不同100ml量瓶中,用水溶解并稀释至刻度,摇匀即得各组分对照品溶液(50μg/ml);取l100-55、peg6000、pvpk30各5mg,置同一100ml量瓶中,用水溶解并稀释至刻度,摇匀即得对照品溶液(50μg/ml)
(2)取peg600020mg,置20ml量瓶中,用水溶解并稀释至刻度,摇匀即得,1mg/ml样品溶液;取1mg/ml样品溶液5ml,置20ml量瓶中,用水溶解并稀释至刻度,摇匀即得,250μg/ml样品溶液;取1mg/ml样品溶液2ml,置20ml量瓶中,用水溶解并稀释至刻度,摇匀即得,100μg/ml样品溶液。
(3)精密量取对照品溶液及样品溶液1.0ml,加入0.5mol/l髙氯酸溶液5.0ml,混匀,室温放置15分钟,以每分钟4000转离心10分钟。取上清液4ml,加人氯化钡溶液(称取氯化钡5g,加水溶解至100ml)1.0ml和0.1mol/l碘溶液(称取碘化钾2.0g,加少量水溶解,然后加碘1.3g,再加水至50ml,摇匀)0.5ml,混匀,室温反应15分钟,照紫外-可见分光光度法(chp2015通则0401),在波长535nm处测定吸光度。
实验结果显示:上述实验步骤(2),1mg/ml、250μg/ml、100μg/mlpeg6000样品溶液加入0.1mol/l碘溶液后,产生沉淀,且浓度越大,产生沉淀越多,无法进行吸光度测定,故判断该方法线性范围较窄;另pvpk30、l100-55在波长535nm处也有吸收,故判断该方法专属性较差,易受其他组分干扰。即该方法无法实现多组分同步进行分离、定量分析。
- 还没有人留言评论。精彩留言会获得点赞!