一种中药口服制剂指纹图谱的建立方法及其指纹图谱与流程
本发明涉及药品检测技术领域,具体为一种中药口服制剂指纹图谱的建立方法及其指纹图谱。
背景技术:
清燥救肺汤临床使用范围很广,涉及到咳嗽、咳血、肺瘘、喉痹、皮肤科疾病汗闭症、瘙痒症以及便秘、见水思尿症、瘘症等多种疾病,其中咳嗽、咳血等肺部疾病占主要部分。从现代医学角度,清燥救肺汤可用于内科、五官科、皮肤科等临床各科,其中最常见的是治疗呼吸道疾病,现已经有一种清燥救肺汤,对其症状提出处方,但是由霜桑叶、石膏、麦冬、炒黑芝麻、人参、阿胶、蜜枇杷叶、甘草、炒苦杏仁九味中药制成,系用水煎煮工艺制成的复方制剂。
原有的方法为:一种清燥救肺汤制剂,由霜桑叶、石膏、麦冬、炒黑芝麻、人参、阿胶、蜜枇杷叶、甘草、炒苦杏仁九味中药制成,系用水煎煮工艺制成的复方制剂,由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣,作为衡量物质基准对应实物(干膏粉)是否与物质基准基本一致的标准参照物,其质量应加强专属性鉴别和多成分、整体质量控制。因此物质基准质量控制要重视以指纹图谱为主的整体质量控制。但本品不同药味成分化学性质差异较大,一个指纹图谱指认的药味相对较少,但是由于中药材的质量良莠不齐,加之成分复杂,现有分析方法很难全面反应药材质量,目前市场上的清燥救肺汤的药品标准中含量基本上只控制一个或两个指标性成分,难以更好的控制药品质量,由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣,作为衡量物质基准对应实物(干膏粉)是否与物质基准基本一致的标准参照物,其质量应加强专属性鉴别和多成分、整体质量控制。因此物质基准质量控制要重视以指纹图谱为主的整体质量控制。但本品不同药味成分化学性质差异较大,一个指纹图谱指认的药味相对较少,故考虑建立两个指纹图谱方法。
综上所述,本发明提供一种中药口服制剂制剂指纹图谱的建立方法及其指纹图谱。采用指纹图谱的建立方法建立的指纹图谱,对清燥救肺汤处方中的桑叶、甘草、人参、炒黑芝麻、蜜枇杷叶五味药材信息进行检查。
技术实现要素:
本发明的目的在于提供一种中药口服制剂指纹图谱的建立方法及其指纹图谱,以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:
一种中药口服制剂指纹图谱的建立方法及其指纹图谱,包括以下步骤:
s1,供试品溶液制备:选用不同批次的清燥救肺汤颗粒样品,精密称定,置容器中,精密加水,超声溶解,放冷,滤过,滤液用乙酸乙酯提取,合并乙酸乙酯,蒸干,残渣加甲醇溶液并转移至量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;
s2,对照品溶液的制备:取甘草苷、咖啡酸、异斛皮苷、异甘草苷加甲醇溶解,制成每1ml含各对照品50μg的溶液,作为对照品溶液;
s3,分别精密吸取参照物溶液和供试品溶液各10μl,注入超高效液相色谱仪,记录90分钟内的指纹图谱,指纹图谱测定的色谱条件为:色谱柱:以十八烷基硅烷键合硅胶为填充剂;柱温:25~35℃;流动相流速:0.8-1.2ml/min;进样量为5-10μl;紫外检测波长:280-360nm;
s4,参照物溶液的制备:取人参皂苷rg1、人参皂苷re、人参皂苷rb1对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得;
s5,分别精密吸取对照品溶液和供试品溶液10μl,注入液相色谱仪,测定,记录85分钟内的指纹图谱,指纹图谱测定的色谱条件为:色谱柱:以十八烷基硅烷键合硅胶为填充剂;柱温:22~28℃;流速:0.8~1.2ml/min;柱温:22~28℃,蒸发光散射检测器检测,蒸发温度为60~80℃,雾化温度为40~60℃,气体流速1.0~1.4l/min,流动相:以乙腈为流动相a,水为流动相b;
s6,将待测样品的指纹图谱积分信号导入中国药典委的“中药色谱指纹图谱相似度评价系统a版”软件;选择不同批次中药口服制剂制剂的指纹图谱中均存在的色谱峰作为共有峰;用平均值计算法计算相似度结果,并生成标准对照指纹图谱;计算各共有峰的相对保留时间;
优选的,所述s1,中药口服制剂颗粒选为制备过程如下:取霜桑叶12份,石膏10份,麦冬5份,炒黑芝麻、甘草各4份,人参、阿胶、蜜枇杷叶、炒苦杏仁各3份,先将炒黑芝麻、炒苦杏仁和人参捣碎,再与剩余除阿胶之外的其它药材加7~12倍水进行煎煮提取,提取时间为1~2小时,提取1~3次,将提取物与阿胶烊化液混合均匀后,进行冷冻干燥制得中药口服制剂冻干粉,备用。
优选的,所述s1,供试品溶液制备具体过程为:取不同批次的中药口服制剂制剂样品,取1g,精密称定,置具塞锥形瓶中,精密加水20ml,超声处理(功率250w,频率40khz)10分钟,放冷,滤过,滤液用乙酸乙酯振摇提取2次,每20ml,合并乙酸乙酯,蒸干,残渣加甲醇溶液并转移至5ml量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
优选的,所述s3,流动相是以下①至④中的任意一种:
①流动相a乙腈–流动相b体积百分浓度0.1%磷酸水溶液
②流动相a乙腈–流动相b体积百分浓度0.2%甲酸水溶液
③流动相a乙腈–流动相b体积百分浓度0.2%醋酸水溶液
④流动相a乙腈–流动相b体积百分浓度0.1%三氟乙酸水溶液
所述s3,色谱条件中洗脱方式采用梯度洗脱,梯度洗脱的流程是以下①至③中的任意一种:
①0~10min,体积百分浓度5-10%流动相a,10~30min,体积百分浓度15-17%流动相a,30~40min,体积百分浓度17%–23%流动相a,40~60min,体积百分浓度23%流动相a,60~65min,体积百分浓度23-42%流动相a,65~90min,体积百分浓度42-50%流动相a。
②0~5min,体积百分浓度10-15%流动相a,5~20min,体积百分浓度15%-20流动相a,20~40min,体积百分浓度20%-30%流动相a,40~60min,体积百分浓度30-%35%流动相a,60~90min,体积百分浓度35-50%流动相a;
③0~10min,体积百分浓度5-15%流动相a,10~30min,体积百分浓度15-30%流动相a,30~40min,体积百分浓度30%流动相a,40~60min,体积百分浓度30%-35%流动相a,60~65min,体积百分浓度35-45%流动相a,65~90min,体积百分浓度45-50%流动相a。
紫外检测波长为280nm、330nm或360nm中的一种。
优选的,所述s4,参照物溶液的制备具体过程为:取本品1g,精密称定,置具塞锥形瓶中,加水50ml,超声处理5~10分钟,用水饱和正丁醇振摇提取2~5次,合并正丁醇液,蒸干,残渣加甲醇溶液并转移至5ml量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
优选的,所述s5,流动相是以下①至④中的任意一种:
①流动相a乙腈–流动相b水溶液
②流动相a乙腈–流动相b体积百分浓度0.2%甲酸水溶液
③流动相a乙腈–流动相b体积百分浓度0.2%醋酸水溶液
④流动相a乙腈–流动相b体积百分浓度0.1%磷酸水溶液
所述s5,色谱条件中洗脱方式采用梯度洗脱,梯度洗脱的流程是以下①至③中的任意一种:
①0~25min,体积百分浓度21%流动相a,25~35min,体积百分浓度21~26%流动相a,35~45min,体积百分浓度26-29%流动相a,45~65min,体积百分浓度29-33%流动相a,65~75min,体积百分浓度33-60%流动相a,75~85min,体积百分浓度60-74%流动相a。
②0~25min,体积百分浓度19%流动相a,25~40min,体积百分浓度19-26%流动相a,40~45min,体积百分浓度26-29%流动相a,45~67min,体积百分浓度29-33%流动相a,67~72min,体积百分浓度33-63%流动相a,72~85min,体积百分浓度63-74%流动相a。
③0~20min,体积百分浓度22%流动相a,20~40min,体积百分浓度22-29%流动相a,40~45min,体积百分浓度29%流动相a,45~65min,体积百分浓度29-33%流动相a,65~72min,体积百分浓度33-62%流动相a,72~85min,体积百分浓度62-74%流动相a。
与现有技术相比,本发明的有益效果是:
本发明中,通过采用指纹图谱的建立方法建立的指纹图谱i和指纹图谱ii,能够全面表征处方中的桑叶、甘草、人参、炒黑芝麻、蜜枇杷叶五味药材信息,更为全面的体现药品质量,从而避免了由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣以及由于中药材的质量良莠不齐,加之成分复杂,分析方法很难全面反应药材质量,清燥救肺汤的药品标准中含量基本上只控制一个或两个指标性成分,难以更好的控制药品质量的问题以及由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣,作为衡量物质基准对应实物(干膏粉)是否与物质基准基本一致的标准参照物,其质量应加强专属性鉴别和多成分、整体质量控制,不同药味成分化学性质差异较大,一个指纹图谱指认的药味相对较少的问题。
附图说明
图1为本发明清燥救肺汤指纹图谱i对照指纹图谱结构示意图;
图2为本发明指纹图谱i-15批中药口服制剂指纹图谱结构示意图;
图3为本发明清燥救肺汤指纹图谱ii对照指纹图谱结构示意图;
图4为本发明指纹图谱ii-15批中药口服制剂指纹图谱结构示意图;
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
请参阅图1-4,本发明提供一种技术方案:
一种中药口服制剂指纹图谱的建立方法及其指纹图谱,包括以下步骤:
s1,供试品溶液制备:选用不同批次的指颗粒样品,精密称定,置容器中,精密加水,超声溶解,放冷,滤过,滤液用乙酸乙酯提取,合并乙酸乙酯,蒸干,残渣加甲醇溶液并转移至量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;
s2,对照品溶液的制备:取甘草苷、咖啡酸、异斛皮苷、异甘草苷加甲醇溶解,制成每1ml含各对照品50μg的溶液,作为对照品溶液;
s3,分别精密吸取参照物溶液和供试品溶液各10μl,注入超高效液相色谱仪,记录90分钟内的指纹图谱,指纹图谱测定的色谱条件为:色谱柱:以十八烷基硅烷键合硅胶为填充剂;柱温:25~35℃;流动相流速:0.8-1.2ml/min;进样量为5-10μl;紫外检测波长:280-360nm;
s4,参照物溶液的制备:取人参皂苷rg1、人参皂苷re、人参皂苷rb1对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得;
s5,分别精密吸取对照品溶液和供试品溶液10μl,注入液相色谱仪,测定,记录85分钟内的指纹图谱,指纹图谱测定的色谱条件为:色谱柱:以十八烷基硅烷键合硅胶为填充剂;柱温:22~28℃;流速:0.8~1.2ml/min;柱温:22~28℃,蒸发光散射检测器检测,蒸发温度为60~80℃,雾化温度为40~60℃,气体流速1.0~1.4l/min,流动相:以乙腈为流动相a,水为流动相b;
s6,将待测样品的指纹图谱积分信号导入中国药典委的“中药色谱指纹图谱相似度评价系统a版”软件;选择不同批次中药口服制剂制剂的指纹图谱中均存在的色谱峰作为共有峰;用平均值计算法计算相似度结果,并生成标准对照指纹图谱;计算各共有峰的相对保留时间;
所述s1,清燥救肺汤颗粒选为制备过程如下:取霜桑叶12份,石膏10份,麦冬5份,炒黑芝麻、甘草各4份,人参、阿胶、蜜枇杷叶、炒苦杏仁各3份,先将炒黑芝麻、炒苦杏仁和人参捣碎,再与剩余除阿胶之外的其它药材加7~12倍水进行煎煮提取,提取时间为1~2小时,提取1~3次,将提取物与阿胶烊化液混合均匀后,进行冷冻干燥制得中药口服制剂冻干粉,备用。
所述s1,供试品溶液制备具体过程为:取不同批次的中药口服制剂制剂样品,取1g,精密称定,置具塞锥形瓶中,精密加水20ml,超声处理(功率250w,频率40khz)10分钟,放冷,滤过,滤液用乙酸乙酯振摇提取2次,每20ml,合并乙酸乙酯,蒸干,残渣加甲醇溶液并转移至5ml量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
所述s3,流动相是以下①至④中的任意一种:
①流动相a乙腈–流动相b体积百分浓度0.1%磷酸水溶液
②流动相a乙腈–流动相b体积百分浓度0.2%甲酸水溶液
③流动相a乙腈–流动相b体积百分浓度0.2%醋酸水溶液
④流动相a乙腈–流动相b体积百分浓度0.1%三氟乙酸水溶液
所述s3,色谱条件中洗脱方式采用梯度洗脱,梯度洗脱的流程是以下①至③中的任意一种:
①0~10min,体积百分浓度5-10%流动相a,10~30min,体积百分浓度15-17%流动相a,30~40min,体积百分浓度17%–23%流动相a,40~60min,体积百分浓度23%流动相a,60~65min,体积百分浓度23-42%流动相a,65~90min,体积百分浓度42-50%流动相a。
②0~5min,体积百分浓度10-15%流动相a,5~20min,体积百分浓度15%-20流动相a,20~40min,体积百分浓度20%-30%流动相a,40~60min,体积百分浓度30-%35%流动相a,60~90min,体积百分浓度35-50%流动相a;
③0~10min,体积百分浓度5-15%流动相a,10~30min,体积百分浓度15-30%流动相a,30~40min,体积百分浓度30%流动相a,40~60min,体积百分浓度30%-35%流动相a,60~65min,体积百分浓度35-45%流动相a,65~90min,体积百分浓度45-50%流动相a。
紫外检测波长为280nm、330nm或360nm中的一种。
所述s4,参照物溶液的制备具体过程为:取本品1g,精密称定,置具塞锥形瓶中,加水50ml,超声处理5~10分钟,用水饱和正丁醇振摇提取2~5次,合并正丁醇液,蒸干,残渣加甲醇溶液并转移至5ml量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
所述s5,流动相是以下①至④中的任意一种:
①流动相a乙腈–流动相b水溶液
②流动相a乙腈–流动相b体积百分浓度0.2%甲酸水溶液
③流动相a乙腈–流动相b体积百分浓度0.2%醋酸水溶液
④流动相a乙腈–流动相b体积百分浓度0.1%磷酸水溶液
所述s5,所述的色谱条件中洗脱方式采用梯度洗脱,梯度洗脱的流程是以下①至③中的任意一种:
①0~25min,体积百分浓度21%流动相a,25~35min,体积百分浓度21~26%流动相a,35~45min,体积百分浓度26-29%流动相a,45~65min,体积百分浓度29-33%流动相a,65~75min,体积百分浓度33-60%流动相a,75~85min,体积百分浓度60-74%流动相a。
②0~25min,体积百分浓度19%流动相a,25~40min,体积百分浓度19-26%流动相a,40~45min,体积百分浓度26-29%流动相a,45~67min,体积百分浓度29-33%流动相a,67~72min,体积百分浓度33-63%流动相a,72~85min,体积百分浓度63-74%流动相a。
③0~20min,体积百分浓度22%流动相a,20~40min,体积百分浓度22-29%流动相a,40~45min,体积百分浓度29%流动相a,45~65min,体积百分浓度29-33%流动相a,65~72min,体积百分浓度33-62%流动相a,72~85min,体积百分浓度62-74%流动相a。
具体实施方法:指纹图谱i归属了桑叶、甘草,指纹图谱ii归属了人参、炒黑芝麻、蜜枇杷叶。
方法1:本发明所述的清燥救肺汤指纹图谱i的建立方法,具体包括以下步骤:
(1)供试品溶液制备:
提供不同批次的清燥救肺汤颗粒样品,精密称定,置容器中,精密加水,超声溶解,放冷,滤过,滤液用乙酸乙酯提取,合并乙酸乙酯,蒸干,残渣加甲醇溶液并转移至量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
(2)对照品溶液的制备:
取甘草苷、咖啡酸、异斛皮苷、异甘草苷加甲醇溶解,制成每1ml含各对照品50μg的溶液,作为对照品溶液。
(3)分别精密吸取参照物溶液和供试品溶液各10μl,注入超高效液相色谱仪,记录90分钟内的指纹图谱,指纹图谱测定的色谱条件为:色谱柱:以十八烷基硅烷键合硅胶为填充剂;柱温:25~35℃;流动相流速:0.8-1.2ml/min;进样量为5-10μl;紫外检测波长:280-360nm。
(4)将待测样品的指纹图谱积分信号导入中国药典委的“中药色谱指纹图谱相似度评价系统a版”软件;选择不同批次清燥救肺汤制剂的指纹图谱中均存在的色谱峰作为共有峰;用平均值计算法计算相似度结果,并生成标准对照指纹图谱;计算各共有峰的相对保留时间。
其中:
步骤(1)所述的清燥救肺汤颗粒的制备过程如下:取霜桑叶12份,石膏10份,麦冬5份,炒黑芝麻、甘草各4份,人参、阿胶、蜜枇杷叶、炒苦杏仁各3份。先将炒黑芝麻、炒苦杏仁和人参捣碎,再与剩余除阿胶之外的其它药材加7~12倍水进行煎煮提取,提取时间为1~2小时,提取1~3次,将提取物与阿胶烊化液混合均匀后,进行冷冻干燥制得中药口服制剂冻干粉,备用。
步骤(1)的具体过程为:提供不同批次的清燥救肺汤制剂样品,取1g,精密称定,置具塞锥形瓶中,精密加水20ml,超声处理(功率250w,频率40khz)10分钟,放冷,滤过,滤液用乙酸乙酯振摇提取2次,每20ml,合并乙酸乙酯,蒸干,残渣加甲醇溶液并转移至5ml量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
步骤(3)所述的流动相是以下①至④中的任意一种:
①流动相a乙腈–流动相b体积百分浓度0.1%磷酸水溶液
②流动相a乙腈–流动相b体积百分浓度0.2%甲酸水溶液
③流动相a乙腈–流动相b体积百分浓度0.2%醋酸水溶液
④流动相a乙腈–流动相b体积百分浓度0.1%三氟乙酸水溶液
步骤(3)所述的色谱条件中洗脱方式采用梯度洗脱,梯度洗脱的流程是以下①至③中的任意一种:
①0~10min,体积百分浓度5-10%流动相a,10~30min,体积百分浓度15-17%流动相a,30~40min,体积百分浓度17%–23%流动相a,40~60min,体积百分浓度23%流动相a,60~65min,体积百分浓度23-42%流动相a,65~90min,体积百分浓度42-50%流动相a。
②0~5min,体积百分浓度10-15%流动相a,5~20min,体积百分浓度15%-20流动相a,20~40min,体积百分浓度20%-30%流动相a,40~60min,体积百分浓度30-%35%流动相a,60~90min,体积百分浓度35-50%流动相a;
③0~10min,体积百分浓度5-15%流动相a,10~30min,体积百分浓度15-30%流动相a,30~40min,体积百分浓度30%流动相a,40~60min,体积百分浓度30%-35%流动相a,60~65min,体积百分浓度35-45%流动相a,65~90min,体积百分浓度45-50%流动相a。
紫外检测波长为280nm、330nm或360nm中的一种。
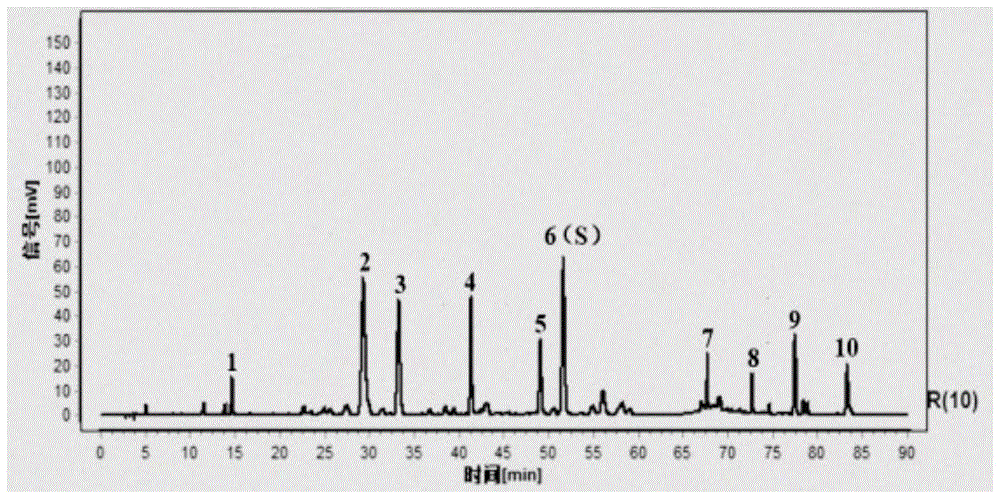
按照上述方法对15批清燥救肺汤颗粒进行指纹图谱测定及分析,选择15批物质基准对应实物指纹图谱中稳定性较好、响应值适宜的色谱法为共有峰,共标定了10个共有峰。采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价软件系统2012年版》,以15批中药口服制剂物质基准对应实物色谱法自动匹配,形成共有峰模式图,建立对照图谱,见附图1。
本发明所述的清燥救肺汤制剂指纹图谱,包括10个共有峰,各共有峰相对保留时间与参照峰相对保留时间比值。分别为:1号峰相对保留时间0.281;2号峰相对保留时间0.550;3号峰相对保留时间0.618;4号峰相对保留时间0.801;5号峰相对保留时间0.953;6号峰相对保留时间1.000;7号峰相对保留时间1.356;8号峰相对保留时间1.454;9号峰相对保留时间1.548;10号峰相对保留时间1.661。其中6号峰为参照峰。
共有峰中,1号峰为咖啡酸,2号峰为甘草苷,3号峰为异槲皮苷,4号峰为紫云英苷,5号峰为芹糖异甘草苷,7号峰为甘草糖苷b和甘草糖苷c,8号峰为5,7-二羟基黄酮。6号峰(s)为异甘草苷。
按照确定的方法对15批供试品进行测定,并对所得指纹图谱进行分析,选择15批样品指纹图谱中稳定性较好、响应值适宜的色谱峰为共有峰,共标定了10个共有峰。见附图2。
方法2:本发明所述的清燥救肺汤制剂指纹图谱ⅱ的建立方法,具体包括以下步骤:
(1)供试品溶液制备:
取不同批次的清燥救肺汤颗粒样品,精密称定,置容器中,精密加水,超声处理一定时间,用水饱和正丁醇振摇提取,合并正丁醇液,蒸干,残渣加甲醇溶液并转移至量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
(2)参照物溶液的制备:
取人参皂苷rg1、人参皂苷re、人参皂苷rb1对照品适量,精密称定,加甲醇制成每1ml含0.1mg的溶液,即得。
(3)分别精密吸取对照品溶液和供试品溶液10μl,注入液相色谱仪,测定,记录85分钟内的指纹图谱,指纹图谱测定的色谱条件为:色谱柱:以十八烷基硅烷键合硅胶为填充剂;柱温:22~28℃;流速:0.8~1.2ml/min;柱温:22~28℃,蒸发光散射检测器检测,蒸发温度为60~80℃,雾化温度为40~60℃,气体流速1.0~1.4l/min,流动相:以乙腈为流动相a,水为流动相b。
(4)将待测样品的指纹图谱积分信号导入中国药典委的“中药色谱指纹图谱相似度评价系统a版”软件;选择不同批次清燥救肺汤制剂的指纹图谱中均存在的色谱峰作为共有峰;用平均值计算法计算相似度结果,并生成标准对照指纹图谱;计算各共有峰的相对保留时间。
其中:
步骤(1)所述的清燥救肺汤颗粒的制备过程如下:取霜桑叶12份,石膏10份,麦冬5份,炒黑芝麻、甘草各4份,人参、阿胶、蜜枇杷叶、炒苦杏仁各3份。先将炒黑芝麻、炒苦杏仁和人参捣碎,再与剩余除阿胶之外的其它药材加7~12倍水进行煎煮提取,提取时间为1~2小时,提取1~3次,将提取物与阿胶烊化液混合均匀后,进行冷冻干燥制得中药口服制剂冻干粉,备用。
步骤(1)的具体过程为:取本品1g,精密称定,置具塞锥形瓶中,加水50ml,超声处理5~10分钟,用水饱和正丁醇振摇提取2~5次,合并正丁醇液,蒸干,残渣加甲醇溶液并转移至5ml量瓶,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
步骤(3)所述的流动相是以下①至④中的任意一种:
①流动相a乙腈–流动相b水溶液
②流动相a乙腈–流动相b体积百分浓度0.2%甲酸水溶液
③流动相a乙腈–流动相b体积百分浓度0.2%醋酸水溶液
④流动相a乙腈–流动相b体积百分浓度0.1%磷酸水溶液
步骤(3)所述的色谱条件中洗脱方式采用梯度洗脱,梯度洗脱的流程是以下①至③中的任意一种:
①0~25min,体积百分浓度21%流动相a,25~35min,体积百分浓度21~26%流动相a,35~45min,体积百分浓度26-29%流动相a,45~65min,体积百分浓度29-33%流动相a,65~75min,体积百分浓度33-60%流动相a,75~85min,体积百分浓度60-74%流动相a。
②0~25min,体积百分浓度19%流动相a,25~40min,体积百分浓度19-26%流动相a,40~45min,体积百分浓度26-29%流动相a,45~67min,体积百分浓度29-33%流动相a,67~72min,体积百分浓度33-63%流动相a,72~85min,体积百分浓度63-74%流动相a。
③0~20min,体积百分浓度22%流动相a,20~40min,体积百分浓度22-29%流动相a,40~45min,体积百分浓度29%流动相a,45~65min,体积百分浓度29-33%流动相a,65~72min,体积百分浓度33-62%流动相a,72~85min,体积百分浓度62-74%流动相a。
按照上述方法对15批清燥救肺汤颗粒进行指纹图谱测定及分析,选择15批物质基准对应实物指纹图谱中稳定性较好、响应值适宜的色谱法为共有峰,共标定了9个共有峰。采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价软件系统2012年版》,以15批清燥救肺汤物质基准对应实物色谱法自动匹配,形成共有峰模式图,建立对照图谱,见附图3。
本发明所述的清燥救肺汤制剂指纹图谱,包括9个共有峰,各共有峰相对保留时间与参照峰相对保留时间比值。分别为:1号峰相对保留时间0.984;2号峰相对保留时间1.000;3号峰相对保留时间1.558;4号峰相对保留时间1.607;5号峰相对保留时间1.692;6号峰相对保留时间1.765;7号峰相对保留时间1.787;8号峰相对保留时间1.953;9号峰相对保留时间1.999;其中2号峰为参照峰。
共有峰中,1号峰为人参皂苷rg1,2号峰(s)为人参皂苷re,3号峰为nerolidol-3-o-{α-l-rhamnopyranosyl-(14)-α-l-rhamnopyranosyl-(12)-[α-l-rhamnopyranosyl-(16)]-β-d-glucopyranoside},4号峰为人参皂苷rb1,5号峰为人参皂苷rb2,6号峰为人参皂苷rb3或人参皂苷rc,7号峰为人参皂苷rd。2号峰(s)为人参皂苷re。
按照确定的方法对15批供试品进行测定,并对所得指纹图谱进行分析,选择15批样品指纹图谱中稳定性较好、响应值适宜的色谱峰为共有峰,共标定了9个共有峰。见附图4。
按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90,见表2。
按中药色谱指纹图谱相似度评价系统,以共有峰计算相似度,供试品指纹图谱与对照指纹图谱的相似度不得低于0.90,见表1。
表1
表2
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!