检测食用油中乙基麦芽酚的方法与流程
1.本发明属于食品添加剂的分析和检测领域,具体而言,涉及一种食用油中可能含有或添加的化学物质的分析方法,更具体而言涉及植物油中乙基麦芽酚的定性和定量检测方法。
背景技术:
2.乙基麦芽酚(cas no:4940-11-8),通常为白色针状或白色结晶粉末。作为一种香味改良剂、增香剂,应用越来越广泛,是烟草、食品、饮料、香精、果酒、日用化妆品等良好的香味增效剂,对食品的香味改善和增强具有显著效果,对甜食起着增甜作用。
3.其作为食品用香料,目前未曾发现乙基麦芽酚存在于天然物质中,它必须通过化学合成的方法得到,因此属于人工合成的一种香精香料。虽然,乙基麦芽酚具有一定的安全性,并且是gb 2760-2014《食品安全国家标准食品添加剂使用标准》允许使用的食品用合成香料,可用于烟草、食品、饮料、香精、酒类等的增香。但gb 2760-2014附录b.1中明确规定植物油脂中不得添加食品用香料、香精。因此,乙基麦芽酚在食用植物油中实际上属于非法添加类,并不得检出。
4.进一步,已经存在对于食品类物质中乙基麦芽酚含量的检测方法,目前检测此类化合物的方法主要集中在紫外分光法、液相色谱法、气相色谱法和液质联用法,以上方法还经常结合固相萃取法一起使用,起到净化样品基质,提高检测准确度的目的。例如:
5.目前gb 5009.250-2016《食品安全国家标准-食品中乙基麦芽酚的测定》规定对食品中乙基麦芽酚的检测方法为高效液相色谱法,此方法的检出限为0.5mg/kg,但该方法的适用范围中并未包括食用油脂产品;
6.已经出台的食品补充检测方法bjs(2017.08,中国)《食用植物油中乙基麦芽酚的测定》,用于食用油脂中乙基麦芽酚的监督抽检项目;此方法为高效液相色谱-三重四级杆质谱联用法;方法检出限为25μg/kg,该方法所采用的仪器设备昂贵,不能得以广泛使用及推广;因油脂样品成分复杂,存在本底物质干扰检测的情况。
7.目前,对于使用气相色谱与质谱联用的对乙基麦芽酚的检测方法,例如:
8.引用文献1公开了一种以固相萃取为提取分离方式的气质联用测定调和油中麦芽酚、乙基麦芽酚的方法。其样品经正己烷溶解后,采用氨基固相萃取柱分离净化,结合气质的高分辨率和高灵敏度,通过选择合适的监测离子,分别从检出限、回收率和精密度3个方面考察方法的适用性。检测结果中,乙基麦芽酚检出限为0.35mg/kg。在0-0.5g/kg测定范围内,加标回收率为95.2%-107%,精密度为0.9%-1.2%。
9.引用文献2公开了一种掺伪花生油的鉴别方法,其包括:萃取预处理以及利用气质联用仪对萃取出来的芳香性物质进行分析,并通过采用xcalibur软件处理实验图谱得到相关数据,利用乙基麦芽酚和三乙酸甘油酯作为特征性物质对花生香精进行定性分析。
10.然而,引用文献1,样品需要经过固相萃取然后进行气质联用测试,其检测的便捷性以及对于乙基麦芽酚的检测精度(检出限)仍然不能说是充分的,引用文献2中仅仅涉及
了定性分析而没有公开定量检测的可行性。
11.此外,在针对其他的蔬菜农残的检测方面中,也存在使用气相色谱与质谱联用的检测手段已对目标农药残留物进行定量检测。
12.例如,引用文献3以常见的4种蔬菜为材料,应用气相色谱/质谱联用方法,研究了色谱柱极性与扫描方式对测定蔬菜中5种菊酯类农药残留量的影响。使用较强极性的毛细管柱(如db-17ms)可降低蔬菜中菊酯类农药的检出限。在确定每种菊酯类农药特征离子的基础上,采用选择离子监控扫描(sim),可显著降低蔬菜中菊酯类农药的检出限,4种蔬菜中5种菊酯类检出限范围在0.00253-0.19100mg/kg。虽然引用文献3具有较低的检出限,但对于食用油中是否能进行类似的检测无法得知,尤其是考虑到植物油中的干扰物质大量存在的情况。
13.可见,对于食用油,尤其是植物油中可能存在的乙基麦芽酚的检测而言,还不存在一种经济、有效的定性、定量测试方法,对于油脂中存在的物质干扰因素可能导致的定量不确定性的研究仍不能说是充分的,因此,仍然存在对检测方法进一步改进的需求。
14.引用文献:
15.引用文献1:《中国食品添加剂》,2017年,第6期第209-213页,
16.引用文献2:cn106526031a,
17.引用文献3:气质联用检测蔬菜中菊酯类农药残留量研究,张静等,《安徽农业科学》,2011。
技术实现要素:
18.发明要解决的问题
19.针对现有技术以上存在的问题或不足,本发明所要解决的技术问题在于,提供一种对于食用油、尤其是植物油中可能存在的乙基麦芽酚添加剂的可靠的检测方法,而且能够解决本底物质干扰导致的定性不准及定量不准确的问题,该方法具有低的检测极限以及优良的检测精度,能够进行良好的定性以及定量测量。
20.此外,本发明的另外的目的还在于提供一种食用油中可能存在的乙基麦芽酚的检测方法,其先通过弱极性色谱柱与质谱联用进行定量检测,如果超出目前安全标准中的定量检测极限值或浓度下限值(即安全阈值),则再使用强极性色谱柱与质谱联用进行定量或定性检测(复检)。这样的方法尤其适用于高效地进行大规模、筛查性检测。
21.此外,本发明的检测方法工艺简单、经济便捷,易于大规模工业应用。
22.用于解决问题的方案
23.经过本发明人长期潜心研究,发现通过如下方法能够解决上述技术问题:
24.[1].本发明首先提供了一种食用油中乙基麦芽酚检测的方法,其为一种定量检测方法,包括如下定量的步骤:
[0025]
样品的制备步骤,通过待测食用油制备待测样品,并基于标准加入法制备n个加标样品,所述n为大于等于3的整数,优选地,所述n为3~10的整数;
[0026]
前处理的步骤,对所述待测样品和加标样品中的极性物质进行富集并得检测样品;优选地,所述富集是使用极性溶剂对待测样品和加标样品进行萃取,所述极性溶剂选自乙腈或者甲醇;
[0027]
检测的步骤,将各个所述检测样品通过气相色谱和质谱的联用进行测试,并基于标准加入法制备工作曲线从而确定所述待测样品中乙基麦芽酚含量,
[0028]
其中,所述气相色谱包括强极性色谱柱,所述质谱采用特征选择离子监测扫描(sim)工作模式;优选地,所述强极性色谱柱选自基于聚乙二醇的色谱柱、极性基团改性的聚乙二醇色谱柱或极性基团改性的聚硅氧烷色谱柱。
[0029]
[2].根据[1]所述的方法,其中,所述检测的步骤中,在气相色谱的所述强极性色谱柱之前和/或之后还使用弱极性色谱柱与之串联。
[0030]
[3].根据[1]或[2]所述的方法,其中,所述弱极性色谱柱包括苯基改性的二甲基聚硅氧烷;和/或所述质谱为单四级杆质谱。
[0031]
[4].根据[1]~[3]任一项所述的方法,其中,所述检测的步骤中,气相色谱的升温程序包括,以10℃/min以下的速度升温至不超过100℃,并保温不超过15min;然后再30~40℃/min升温至230~260℃,并保持2~10min。
[0032]
[5].根据[1]~[4]任一项所述的方法,其中,所述方法依据标准加入法工作曲线的线性相关系数r2大于0.998;和/或所述方法对乙基麦芽酚的检出限为5μg/kg。
[0033]
[6].进一步,本发明还提供了一种食用油中乙基麦芽酚检测的方法,其特别是一种简便的定性检测方法,包括如下定性的步骤:
[0034]
样品的制备步骤,通过待测食用油制备待测样品,并制备具有已知乙基麦芽酚浓度的标准样品;
[0035]
前处理的步骤,对所述待测样品中的极性物质进行富集并得检测样品;优选地,所述富集是使用极性溶剂对待测样品进行萃取,所述极性溶剂选自乙腈或者甲醇;
[0036]
检测的步骤,分别将所述标准样品和所述检测样品通过气相色谱和质谱的联用进行测试,并记录:
[0037]
i)标准样品中乙基麦芽酚气相色谱峰的保留时间t1,以及质谱中特征离子峰的丰度比;
[0038]
ii)检测样品中疑似乙基麦芽酚气相色谱峰的保留时间t2,以及质谱中特征离子峰的丰度比,
[0039]
其中,所述气相色谱中包括强极性色谱柱,所述质谱采用特征选择离子监测扫描(sim)工作模式;优选地,所述强极性色谱柱选自基于聚乙二醇的色谱柱、极性基团改性的聚乙二醇色谱柱或极性基团改性的聚硅氧烷色谱柱,和/或所述质谱为单四级杆质谱;
[0040]
判断的步骤,通过t1与t2的比较,并且必要时还通过所述质谱中特征离子峰的丰度比的比较确定待测样品中是否含有乙基麦芽酚,
[0041]
[7].根据[6]所述的方法,其中,所述检测的步骤中,气相色谱的升温程序包括,以10℃/min以下的速度升温至不超过100℃,并保温不超过15min;然后再30~40℃/min升温至230~260℃,并保持2~10min。
[0042]
[8].根据[6]或[7]所述的方法,其中,在所述判断的步骤中,若所述t1与t2偏差小于等于
±
0.05min,则继续通过质谱特征离子峰的丰度比的比较确定待测样品中是否含有乙基麦芽酚。
[0043]
[9].根据[7]或[8]所述的方法,其中,所述质谱中特征离子峰的丰度比的比较,包括将所述检测样品的139,125和97离子峰相对强度与140离子峰相对强度之比分别与标准
样品中139,125和97离子峰相对强度与140离子峰相对强度之比进行比较。
[0044]
一些具体的实施方案中,上述[6]~[9]的方法可以采用如下的步骤:
[0045]
a.配置乙基麦芽酚含量已知的标准样品(200μg/kg的标准样品);
[0046]
b.对该标准样品进行气质联用检测,并记录乙基麦芽酚在气相色谱中的保留时间t1以及质谱中139,125和97离子峰相对强度与140离子峰相对强度(基准丰度比);
[0047]
c.对待测样品进行前处理,得到检测样品,将检测样品在与b相同的条件下进行气质联用检测,并记录乙基麦芽酚在气相色谱中的保留时间t2以及质谱中139,125和97离子峰相对强度与140离子峰相对强度(检测丰度比);
[0048]
d.将t1和t2、基准丰度比和检测丰度比进行对比。
[0049]
[10].进一步,本发明还提供了一种食用油中乙基麦芽酚检测的方法,其包括如下步骤:
[0050]
样品的制备步骤,通过待测食用油制备待测样品,并制备浓度为安全标准阈值浓度的含有乙基麦芽酚的标准样品;
[0051]
前处理的步骤,对所述待测样品中的极性物质进行富集并得检测样品;优选地,所述富集是使用极性溶剂对待测样品进行萃取,所述极性溶剂选自乙腈或者甲醇;
[0052]
检测的步骤,分别将所述检测样品和标准样品通过气相色谱和质谱的联用进行测试,所述气相色谱中使用弱极性色谱柱,所述质谱采用特征选择离子监测扫描(sim)工作模式;优选地,所述弱极性色谱柱选自苯基改性的二甲基聚硅氧烷,和/或所述质谱为单四极杆质谱;
[0053]
判断的步骤,如果所述检测的步骤的结果显示该所述待测样品中乙基麦芽酚的检测浓度大于安全标准阈值浓度,则将气相色谱中的弱极性色谱柱更换为强极性色谱柱或将所述弱极性色谱柱与强极性色谱柱串联,并与所述质谱联用进行测试以对该待测样品重新进行定量或定性检测。
[0054]
[11].根据[10]所述的方法,其中,所述判断的步骤中,如果检测样品中的质谱定量离子140离子所对应的峰面积大于标准样品定量离子140离子所对应的峰面积,则认为该所述待测样品中乙基麦芽酚的检测浓度大于安全标准阈值浓度。
[0055]
[12].根据[10]或[11]所述的方法,其特征在于,所述检测的步骤中,气相色谱的升温程序包括,以3~7℃/min速度升温至不超过160℃,并保温不超过15min;然后再以8~12℃/min升温至190~210℃,并保持0.5~2min;后再25~35℃/min升温至230~255℃,并保持1~3min。
[0056]
[13].根据[10]~[12]任一项所述的方法,其中,所述安全标准阈值浓度为25μg/kg。
[0057]
[14].根据[10]~[13]任一项所述的方法,其中,所述弱极性色谱柱与强极性色谱柱串联方式包括,在强极性色谱柱的前和/或后串联弱极性色谱柱。
[0058]
一些具体的实施方案中,上述[10]~[14]的方法可以采用如下的步骤:
[0059]
a.确定安全标准阈值浓度,并配置该阈值浓度的含有乙基麦芽酚的标准样品(例如25μg/kg的标准样品),并使用气质联用系统进行检测,并记录与质谱定量离子140所对应的色谱峰的峰面积(基准峰面积);
[0060]
b.取待测样品经前处理得到检测样品,并在与a相同的测试条件下进行气质联用
检测,并记录与质谱定量离子140所对应的色谱峰的峰面积(检测峰面积);
[0061]
c.将基准峰面积与检测峰面积对比,如果检测峰面积高于基准峰面积,则将气相色谱中的弱极性色谱柱更换为强极性色谱柱或将所述弱极性色谱柱与强极性色谱柱串联,并与所述质谱联用进行测试以对该待测样品重新进行定量或定性检测。
[0062]
[15].根据[10]~[14]任一项所述的方法,其中,所述重新进行定量或定性检测包括使用以上[1]~[5]任一项所述的检测方法。
[0063]
发明的效果
[0064]
通过上述技术方案的实施,本发明能够获得如下的技术效果:
[0065]
1)本发明采用强极性色谱柱、或弱极性色谱柱与强极性色谱柱串联方法在优选的色谱条件能够使乙基麦芽酚在气相色谱分离过程中避免共流出峰情况,因此可以完全分离乙基麦芽酚及干扰物质。同时,采用特征选择离子监测扫描模式(sim)监测,通过标准加入外标法-气相色谱/质谱联用法,可以准确测定食用油脂中乙基麦芽酚的含量,方法定量准确、灵敏、准确度和重现性好。一些具体的实施方案中,采用强极性色谱柱或使弱极性色谱柱与强极性色谱柱串联方法,乙基麦芽酚峰形良好、对称,不产生拖尾峰现象。
[0066]
2)线性试验及检测限试验结果表明,在一定浓度范围内峰面积与质量浓度的线性关系良好(r2》0.998);
[0067]
3)本发明检测方法的检出限较低,在一些具体的实施方案中,检出限可以低至5μg/kg。
[0068]
4)本发明的检测方法中,具有良好的回收率以及相对标准偏差(rsd)。
[0069]
5)本发明一些具体的实施方案中,采用包括强极性色谱柱的色谱与质谱联用的方法,提供了一种对于待测样品中乙基麦芽酚进行定性检测的方法。
[0070]
6)本发明另外一些优选的实施方案中,采用使用弱极性色谱与质谱联用,如果某个待测样品的定量检测结果超出安全标准阈值浓度,则采用强极性色谱柱复测的方法,有利于大规模待测样品的快速检测和评价。
附图说明
[0071]
图1:本发明实施例中气相色谱-质谱联用的检测系统对标准样品的检测结果;
[0072]
图2:本发明实施例1中通过加标样品制备的工作曲线;
[0073]
图3:本发明实施例1中浓香菜籽油加标样品和待测样品(未加标的实际油样)检测时气相色谱图叠加图;
[0074]
图4:本发明实施例2中精炼四级菜籽油加标样品和待测样品(未加标的实际油样)检测时气相色谱图叠加图;
[0075]
图5:图3与图4的气相色谱叠加图。
具体实施方式
[0076]
以下对本发明的实施方式进行说明,但本发明不限定于此。本发明不限于以下说明的各构成,在发明请求保护的范围内可以进行各种变更,而适当组合不同实施方式、实施例中各自公开的技术手段而得到的实施方式、实施例也包含在本发明的技术范围中。另外,本说明书中记载的文献全部作为参考文献在本说明书中进行援引。
[0077]
除非另有定义,本发明所用的技术和科学术语具有与本发明所属技术领域中的普通技术人员所通常理解的相同含义。
[0078]
本说明书中,使用“数值a~数值b”或“数值a-数值b”表示的数值范围是指包含端点数值a、b的范围。
[0079]
本说明书中,使用“可以”表示的含义包括了进行某种处理以及不进行某种处理两方面的含义。本说明书中,“任选的”或“任选地”是指接下来描述的事件或情况可发生或可不发生,并且该描述包括该事件发生的情况和该事件不发生的情况。
[0080]
本发明中,使用“安全标准”表示与食品质量检测或监测相关的地方、地区、国家、区域或国际组织中存在的对于以及乙基麦芽酚在食品、食用油中的最低检出量的标准。
[0081]
本说明书中,所提及的“一些具体/优选的实施方式”、“另一些具体/优选的实施方式”、“一些具体/优选的技术方案”、“另一些具体/优选的技术方案”等是指所描述的与该实施方式有关的特定要素(例如,特征、结构、性质和/或特性)包括在此处所述的至少一种实施方式中,并且可存在于其它实施方式中或者可不存在于其它实施方式中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方式中。
[0082]
本发明的术语“标准样品”、“加标样品”、“待测样品”、“检测样品”分别具有以下的含义:
[0083]
标准样品:即具有确定目标分析物的含量或浓度的样品,并且该样品中,不含有对目标分析物产生检测干扰的成分,或者是除了非干扰性的必要溶剂和目标分析物以外,不添加其他成分的样品;
[0084]
加标样品:使用标准样品与待测样品混合得到的样品,混合后的样品以加入的标准样品中的目标分析物的量来计算混合后样品中的该目标分析物的浓度,并将该浓度视为加标样品中目标分析物的浓度;
[0085]
待测样品:指的是检测的对象样品,这样的待测样品为实际取样的样品,在本发明中,也指的是实际需要检测的油样;
[0086]
检测样品:指的是实际上机检测的样品,即对加标样品、待测样品经过前处理后得到的样品,该样品可以直接上机检测。
[0087]
本发明的说明书和权利要求书及上述附图中的术语“包括”以及它们任何变形,意图在于覆盖不排他的包含。例如包含一系列步骤或单元的过程、方法或系统、产品或设备没有限定于已列出的步骤或单元,而是可选地还包括没有列出的步骤或单元,或可选地还包括对于这些过程、方法、产品或设备固有的其它步骤或单元。
[0088]
《第一方面》
[0089]
本发明的第一方面中,提供了一种对于食用油、尤其是植物油中可能存在的乙基麦芽酚添加剂的可靠的定量检测方法,而且能够解决本底物质干扰导致的定量不准确的问题,该方法具有低的检测极限以及优良的检测精度,能够进行良好的定量测量。
[0090]
更具体而言,在本发明的第一方面中,基于标准加入法,通过气相色谱和质谱的联用进行测试,并基于标准加入法制备工作曲线从而确定所述待测样品中乙基麦芽酚的含量。
[0091]
乙基麦芽酚和植物油
[0092]
如前所述,乙基麦芽酚是一种食品工业中常用的香味剂,但由于其被禁止用于食
用油中,因此,对于食用油中鉴别是否在其中非法添加了该香味剂而言,可靠而精准的检测方法是必要的。
[0093]
本发明的食用油,在本发明一些具体的实施方案中,通常指各种植物油。
[0094]
对于这些植物油,包括但不限于:稻米油、葵花籽油、棕榈油、棕榈仁油、花生油、菜籽油、大豆油、亚麻籽油、棉籽油、红花籽油、紫苏籽油、茶籽油、草麻籽油、荷荷巴油、橄榄油、可可豆油、乌桕籽油、扁桃仁油、杏仁油、油桐籽油、橡胶籽泊、玉米胚油、小麦胚油、芝麻籽油、月见草籽油、榛子油、南瓜籽油、胡桃油、葡萄籽油、胡麻籽油、玻璃苣籽油、沙棘籽油、番茄籽油、澳洲坚果油和椰子油中的一种或多种。
[0095]
另外,本发明所述的食用油也包括上述各种植物油按照任意比例所形成的混合油或调和油。
[0096]
进一步,对于这些植物油的生产方式,本发明没有特别的限定,可以通过物理压榨或者通过化学浸出的方法而得到。
[0097]
此外,对于本发明食用油,在一些具体的实施方案中,包括未使用过的食用油,在其他一些实施方案中,也可以为使用过或经过高温加热后的食用油。也就是说,本发明所提供的检测方法不仅可以针对新鲜的食用油,也可以适用于高温使用过的食用油。
[0098]
基于标准加入法的气质联用检测
[0099]
本发明的检测方法是基于标准加入法而进行的定量的检测方法。
[0100]
标准加入法,是将具有一定浓度梯度的已知浓度的标准溶液加入待测样品中制备多个加标样品,通过测定这些加标样品(以及空白样品)的某一特性,从而通过拟合的直线制备工作曲线的方法。这种方法尤其适用于检验样品中是否存在干扰物质。
[0101]
在本发明一些具体的实施方案中,通过使用待测食用油对已知浓度的含有乙基麦芽酚的标准样品进行稀释,在得到待测样品的同时,也得到n个加标样品。
[0102]
对于加标样品的数量,没有特别限制,可以理解的是,加标样品的数量越多,将可能提高测试结果的可靠性,但同时也可能对线性拟合带来更复杂的处理过程。在本发明一些具体的实施方案中,n为大于等于3的整数,优选为3~10的整数,进一步优选为4~6整数。
[0103]
进一步,对于加标样品的乙基麦芽酚的浓度,没有特别的限制,但从线性范围以及检测限的方面考虑,在本发明一些优选的实施方案中,加标样品的浓度可以为10μg/kg~500μg/kg,进一步优选为15μg/kg~350μg/kg,更优选为15μg/kg~300μg/kg。例如,这些加标样品的浓度可以为15μg/kg、25μg/kg、30μg/kg、35μg/kg、40μg/kg、45μg/kg、50μg/kg、55μg/kg、80μg/kg、100μg/kg、120μg/kg、140μg/kg、160μg/kg、180μg/kg或200μg/kg等。
[0104]
(前处理的步骤)
[0105]
本发明前处理步骤为对样品,包括待测样品和加标样品中成分的预分离。
[0106]
油脂中,存在大量的极性成分(强极性成分和弱极性成分)以及非极性成分。在本发明一种具体的实施方案中,通过使用溶剂对样品进行萃取以对极性成分和非极性成分进行预分离处理。
[0107]
对于这样的溶剂,可以使用各种极性溶剂,在一些优选的实施方案中,可以使用乙腈或甲醇作为溶剂与各样品进行混合。由于乙腈或甲醇属于强极性溶剂,因此,样品中的极性物质进入到乙腈等强极性溶剂中被萃取。需要注意的是,乙醇、氯仿、dmso因易与油脂互溶,不能用于该前处理步骤。
[0108]
另外,在本发明前处理步骤中,也可使用辅助手段用于油脂中成分的预分离。对于这样的辅助手段,典型地,可以使用离心分离。在一些优选的实施方案中,离心分离处理的转速可以为4000r/min以上,离心时间可以3~8min。
[0109]
通过上述前处理步骤的萃取和离心处理,富集了样品中的极性成分,并分离去除了至少部分或全部的非极性成分,排除了部分本底物质的干扰。
[0110]
(气相色谱)
[0111]
本发明通过气相色谱对前处理得到的富集成分或萃取物进行分离。通过气相色谱控制条件的优化,可以将源自于油脂样品中的极性成分(尤其是强极性成分)与弱极性的乙基麦芽酚进行分离,排除另一部分本底物质的干扰,从而为乙基麦芽酚的定量检测提供可能。
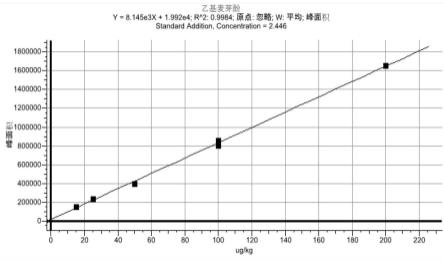
[0112]
对于气相色谱的进样方式,没有特别的限制,可以手动进样,也可以使用自动进样器进样。在一些具体实施方案中,采用自动进样器、不分流进样。
[0113]
本发明的气相色谱中,至少包括一个强极性色谱柱,进而通过所述强极性色谱柱对检测物中的各种极性不同的化学物质进行分离,进而通过后文所述的质谱进行检测。
[0114]
在本发明另外一种具体的实施方案中,在强极性色谱柱之前,还包括一个与该强极性色谱柱串联的弱极性色谱柱,此时,在进行检测时,可以通过弱极性色谱柱对各种极性不同的化学物质进行预分离,预分离后进而通过强极性色谱柱进行再次分离。本发明中,使用弱极性色谱柱与强极性色谱柱串联的方式,有利于将乙基麦芽酚与其他的干扰性成分进行更好的分离。
[0115]
在本发明的另外一种具体的实施方案中,在强极性色谱柱之后,还包括使用一个与该强极性色谱柱串联的弱极性色谱柱。此时,在进行检测时,在使用强极性色谱柱进行成分分离后,继续使用弱极性色谱柱分离。本发明同样认为,这样的色谱柱的连接方式也有利于将乙基麦芽酚与其他的干扰性成分进行更好的分离。
[0116]
另外,对于至少存在一个强极性色谱柱的其他色谱柱设置方式也没有特别的限定,只要不妨碍本发明的技术效果即可。例如,以在强极性色谱柱的前后各设置一个弱极性色谱柱;与强极性色谱柱并联一个弱极性色谱柱等。
[0117]
对于本发明所述的“强极性色谱柱”以及“弱极性色谱柱”可以根据本领域常规的市售色谱柱进行选择。
[0118]
在本发明一些具体的实施方案中,所谓“强极性色谱柱”可以选自基于聚乙二醇的色谱柱、极性基团改性的聚乙二醇色谱柱或极性基团改性的聚硅氧烷色谱柱等。更具体而言,所谓“强极性色谱柱”可以选自商品名为hp-innowax ms、peg20m、bp-10ms、rsl-1701ms、db-1701ms、hp-1701ms和cpisl-19ms的色谱柱。
[0119]
另外,对于所谓“弱极性色谱柱”,可以选自苯基改性的二甲基聚硅氧烷,在一些优选的情况下,苯基的含量不超过8%,优选不超过5%。更具体而言,所谓“弱极性色谱柱”可以选自商品名为:db-5ms;hp-5ms、tg-5ms;se-54ms;rtx-5ms;db-1ms和hp-1ms的色谱柱。
[0120]
通常,对于上述的“强极性色谱柱”以及“弱极性色谱柱”可以从安捷伦、赛默飞世尔等公司商购得到。
[0121]
在使用上述商购的色谱柱进行强极性与弱极性色谱柱进行串联时,在一些具体的实施方案中可以采用玻璃锥形毛细管两通将弱极性色谱柱(如db-5ms)与强极性色谱柱(如
db-1701ms)连接。例如,将弱极性色谱柱的出口插入玻璃锥形毛细管两通入口,将强极性色谱柱一头插入玻璃锥形毛细管两通出口,强极性色谱柱的出口接入质谱检测器。其他的连接方式中,例如将强极性与弱极性色谱柱并联的方式,优选地,可以通过多通阀组来实现检测通路的转换。
[0122]
另外,对于本发明的色谱柱的总长度(包括强极性色谱柱以及可能与之串联连接的一个或前后两个弱极性色谱柱的总长度),从设备的便利性以及检测的可靠性方面考虑,可以为不超过100m,优选为不超过80m,进一步优选为不超过70m。在一个实施例中,弱极性色谱柱与强极性色谱柱串联,色谱柱长度60米,此时,在仪器软件上设置色谱柱长为60米。开启仪器,自检合格后即完成连接。
[0123]
对于气相色谱的操作温度,本发明认为在气相色谱分离中,采用充分的加热,有利于使强极性物质与弱极性的乙基麦芽酚分离。
[0124]
在本发明一些具体的实施方案中,在气相色谱检测中,可以采用分段升温的温控方式。首先,是色谱柱获得一定的初始温度,如50℃以上的初始温度,进一步可以使用较慢的升温速率升温至不超过100℃,并进行不超过15min的保温,这样的升温速率可以为不超过10℃/min;继而,通过较快速的升温速率升温至230~260℃,这样的升温速率可以为30~40℃/min,并在升温终点保温2~10min。
[0125]
在本发明一些优选的实施方案中,本发明的气相色谱可以采用如下的温控方式:将色谱柱初始温度设置为55~65℃,并保持3~5min(例如3.5min、4min等);进而以2.5~5℃/min速度(例如3℃/min、3.5℃/min、4℃/min等)升温至不超过100℃,并保温不超过15min(例如8~13min等);然后再以25~35℃/min速率升温(例如28℃/min、30℃/min、32℃/min等)至230~255℃(例如240℃、250℃等),并保持3~8min(例如5min、7min等)。
[0126]
通过上述的温控方式,使得强极性物质能够更好的渗入强极性色谱柱固相结构中,进而能够将这些物质与弱极性的乙基麦芽酚分离。
[0127]
此外,本发明的气相色谱的载气(流动相)选自惰性气体,例如氦气等。
[0128]
对于气相色谱的其他元件或操作方式没有特别的限制,可以参考本领域常规的技术手段或操作方式。
[0129]
(质谱)
[0130]
本发明使用质谱对气相色谱的分离物进行质谱检测。
[0131]
本发明一些具体的实施方案中,作为质谱检测器,采用特征选择离子监测扫描(sim)工作模式。对于可扫描的特征离子峰,至少包括作为定量离子的140离子峰;对于其他可扫描的离子峰可以包括作为定性离子的139,125以及97离子峰,这主要与质谱检测结果中离子峰的相对峰的强度相关。
[0132]
此外,对于本发明的质谱检测的电离方式,没有特别限制,在一些优选的实施方案中,可以使用电子轰击电离源(ei)进行轰击电离。
[0133]
在本发明一些优选的实施方案中,使用单四极杆质谱。
[0134]
检测可靠性
[0135]
根据本发明所述的气质联用的测试方法,依据标准加入法工作曲线的线性相关系数r2大于0.998,并且所述方法对乙基麦芽酚的检出限为5μg/kg(3倍信噪比计算)。更具体而言:
[0136]
基于强极性色谱柱的测试方法中:乙基麦芽酚含量在10~200μg/kg的范围内线性关系良好,线性方程为y=8.145
×
103x+1.992
×
104,相关系数为0.9984;该方法测得乙基麦芽酚回收率为95-110%,rsd为8%;
[0137]
基于弱极性色谱柱(前)与强极性色谱柱(后)串联法:乙基麦芽酚含量在15~200μg/kg内线性关系良好,线性方程为y=655.57x+7993.07,相关系数为0.9990;该方法测得乙基麦芽酚加标回收率为82-95%,rsd为2.2~9.8%。
[0138]
《第二方面》
[0139]
本发明的第二方面中,提供了一种对于食用油、尤其是植物油中可能存在的乙基麦芽酚添加剂的简便可靠的定性检测方法,而且能够解决本底物质干扰导致的定性不准确的问题。
[0140]
需要说明的是,本发明所提供的定性检测方法可以在第一方面中的定量检测前实施,另外,第一方面的定量检测前可以使用的定性检测方法也并非仅仅局限于本发明该方面所公开的方法。对于这样的检测结果首先要确定最终的质谱检测峰是否代表了乙基麦芽酚的存在,以排出干扰物质的影响。
[0141]
更具体而言,在本发明的第二方面中,使用第一方面中所公开的气相色谱和质谱的联用测试系统和条件,通过将待测样品以及已知浓度的标准样品分别进行气相色谱或者气相色谱与质谱联用的方法进行检测,并判断待测样品中是否含有乙基麦芽酚。
[0142]
首先,通过待测食用油制备待测样品,并制备具有已知(任意)浓度的乙基麦芽酚的标准样品;
[0143]
进一步进行前处理步骤,对所述待测样品中的极性物质进行富集并得检测样品;优选地,所述富集是使用极性溶剂对待测样品进行萃取,所述极性溶剂选自乙腈或者甲醇;
[0144]
定性检测的步骤主要基于以下两点思路而设计:
[0145]
第i)点,通过气相色谱乙基麦芽酚的特征峰的保留时间来进行确定。具体而言:首先采用已知任意浓度的标准样品进行本发明上文所述的气相色谱(或气质联用)检测,确定该标准样品中乙基麦芽酚的保留时间。进而在相同的气相色谱条件下,对待测样品经过前处理得到的检测样品进行检测,如果其气相色谱中疑似乙基麦芽酚峰的保留时间与标准样品的保留时间相同或者偏差小于等于
±
0.05min,则可以进行如下第ii)点的相关定性检测。
[0146]
第ii)点,通过质谱离子峰的丰度比对该样品中是否存在乙基麦芽酚进行(定性)确定。一般而言,对于同样一个气相色谱-质谱联用仪器进行物质检测时,每次检测时,各个特征峰的丰度比是确定的。例如,当使用一种适用于本发明的气相色谱-质谱联用仪器进行检测时,并使用已知不同浓度的样品(也可以是已知浓度的标准样品)进行检测时,虽然浓度不同,包括140离子峰以及139,125和97离子峰的相对强度在每个检测结果中的强度有变化,然而,139离子峰与140离子峰的相对强度之比、125离子峰与140离子峰的相对强度之比、97离子峰与140离子峰的相对强度之比在不同浓度样品的质谱检测结果上是基本相同的。
[0147]
综合以上第i)点和第ii)点考虑,本发明可以采用以下的方法对气相色谱-质谱联用仪器检测的质谱结果是否代表乙基麦芽酚进行定性确定:
[0148]
①
使用已知(任意)浓度的乙基麦芽酚的标准样品,在确定的条件下(所述的确定
的条件可以参考上文本发明第一方面公开的内容),使用气相色谱-质谱联用进行检测,并记录气相色谱中乙基麦芽酚特征峰的保留时间。进而在上述确定的条件下,对待测样品经前处理后的检测样品进行检测,如果检测样品的气相色谱特征峰的保留时间与标准样品的保留时间相同或者偏差小于等于
±
0.05min,则可以进行以下进一步定性检测;
[0149]
②
使用已知(任意)浓度的含有乙基麦芽酚的标准样品(可以与上文i)中使用的标准样品相同),在确定的条件下(所述的确定的条件可以参考上文本发明第一方面公开的内容),使用气相色谱-质谱联用进行检测,并得到该标准样品的139离子峰与140离子峰的相对强度之比、125离子峰与140离子峰的相对强度之比、97离子峰与140离子峰的相对强度之比,作为定性对照标准。此处的相对强度之比与上文所述的丰度比具有等同的物理意义。进一步在相同的条件下对待测样品经前处理后的检测样品进行气质联用检测,得到质谱检测结果,确定该结果中139,125和97离子峰相对强度与140离子峰相对强度之比,并将该离子丰度比与上述定性对照标准进行对比,如果各个离子丰度比与定性对照标准的偏差满足以下要求(参见下表1),则说明待测样品中确定含有乙基麦芽酚。
[0150]
表1离子丰度比最大允许偏差(与标准样品离子丰度比较)
[0151]
丰度比(%,以140离子峰为基峰)允许偏差>50
±
10%20~50
±
15%10~20
±
20%≤10
±
50%
[0152]
需要说明的是,在一些优选的实施方案中,步骤
②
可以与步骤
①
在一次测量中全部进行。例如,对于上述已知(任意)浓度的含有乙基麦芽酚的标准样品,可以在确定的条件下进行使用气相色谱-质谱联用进行检测,并记录气相色谱中乙基麦芽酚特征峰的保留时间以及记录质谱各个离子的标准丰度比。对于待测样品也可以如此进行,经前处理后得到检测样品后,在相同的条件下,对检测样品进行气相色谱-质谱联用检测,得到保留时间和各个离子的丰度比。进而参照上文所述方法将待测样品保留时间和各个离子丰度比与标准样品进行相应的对比,以确定待测样品中是否含有乙基麦芽酚或者是确定将要进行定量分析的对象是否为乙基麦芽酚。
[0153]
总之,本发明第二方面中所提供的针对食用油中的乙基麦芽酚的气质联用检测方法,是一种简便、经济并且可靠的定性的测试方法,为食品安全检测提供了新的解决方案。
[0154]
《第三方面》
[0155]
本发明的第三方面中,提供了另外一种用于食用油、尤其是植物油中可能存在的乙基麦芽酚添加剂的可靠的检测方法。
[0156]
具体而言,这样的检测方法包括:
[0157]
样品的制备步骤,通过待测食用油制备待测样品,并制备浓度为安全标准阈值浓度的含有乙基麦芽酚的标准样品;
[0158]
前处理的步骤,对所述待测样品中的极性物质进行富集并得检测样品;
[0159]
检测的步骤,分别将所述检测样品和标准样品通过气相色谱和质谱的联用进行测试,所述气相色谱中使用弱极性色谱柱,所述质谱采用特征选择离子监测扫描(sim)工作模式;优选地,所述弱极性色谱柱选自苯基改性的二甲基聚硅氧烷,和/或所述质谱为单四极
杆质谱;
[0160]
判断的步骤,如果所述检测的步骤的结果显示该所述待测样品中乙基麦芽酚的检测浓度大于安全标准阈值浓度,则将气相色谱中的弱极性色谱柱更换为强极性色谱柱或将所述弱极性色谱柱与强极性色谱柱串联,并与所述质谱联用进行测试以对该待测样品重新进行定量或定性检测判断(复检)。
[0161]
在所述的判断的步骤中,在本发明一些具体的实施方案中,(可以在确定的条件下,例如下文将提及的气相色谱和质谱的工作条件)对具有安全标准阈值浓度含量的、含有乙基麦芽酚的标准样品进行气质联用测试,以得到该标准样品的140离子对应的峰的面积;进而对待测样品在相同检测条件下进行检测,得到该待测样品的140离子所对应的峰的面积。如果待测样品的140离子峰的面积不高于上述标准样品,则说明待测样品中可能存在的乙基麦芽酚的含量不高于安全标准阈值浓度,即无需进行使用含有强极性色谱柱的气质联用复检,反之对该样品需要进行定性或定量复检。
[0162]
本发明的第三方面的检测方法主要是基于以下的考虑:
[0163]
在气相色谱与质谱联用检测食用油中可能存在的乙基麦芽酚含量时,当气相色谱中使用弱极性色谱柱时,相对于使用强极性色谱柱的情况而言,具有更快的检测效率。然而,虽然检测效率方面具有优势,但使用弱极性色谱柱对检测样品进行分离时,对于一些干扰性物质与乙基麦芽酚的分离可能存在不充分的担忧。因此,理论上,使用弱极性色谱柱进行气相色谱与质谱联用检测时,定量检测出的检测值通常高于待测样品中实际乙基麦芽酚的含量。
[0164]
根据上述的考虑,本发明提供了特别适用于高效率地(大规模)筛查问题品的方法。该方式先对待测样品进行基于弱极性色谱柱的气相色谱与质谱的联用进行定量检测:
[0165]
i)如果该待测样品的检出值没有超过安全标准阈值浓度,则说明该待测样品满足该安全标准的要求;
[0166]
ii)如果该待测样品的检出值超过安全标准阈值浓度,则说明该待测样品可能存在问题(因为基于弱极性色谱柱的气质联用结果可能无法区分一些干扰物质),在这样的情况下,需要将弱极性色谱柱更换为强极性色谱柱或将所述弱极性色谱柱与强极性色谱柱串联(所述弱极性色谱柱与强极性色谱柱串联方式包括,在强极性色谱柱的前和/或后串联弱极性色谱柱),然后对该待测样品进行气质联用的检测,这样的检测可以为定量检测也可以为定性检测(视具体需要而确定),具体检测方法,如下文所述,仍然可以按照本发明《第一方面》公开的方法进行。
[0167]
因此可见,当本方面的方法面对众多的待测样品,且这些样品中极少存在问题时,可以进行高效的筛查。
[0168]
基于弱极性色谱柱的气质联用检测
[0169]
本方面的检测方法的样品制备步骤以及前处理的步骤,可以参照本发明《第一方面》进行。
[0170]
本方面中的所述弱极性色谱柱可以依照《第一方面》所公开的弱极性色谱柱进行选择。同样,在优选的实施方案中,使用苯基含量不超过8%的苯基改性的二甲基聚硅氧烷。
[0171]
另外,对于使用弱极性色谱柱时的气相色谱的升温方式而言,可以采用分段升温的温控方式。一些具体的实施方案中气相色谱的升温程序包括,以3~7℃/min速度升温至
不超过160℃,并保温不超过15min;然后再以8~12℃/min升温至190~210℃,并保持0.5~2min;后再25~35℃/min升温至230~255℃,并保持1~3min。
[0172]
更具体而言,例如将色谱柱初始柱温设定为58~62℃并保持1~3min;进一步,以3~7℃/min速度(例如4℃/min、5℃/min等)升温至不超过160℃(例如155℃、150℃等),并保温不超过15min(例如7~10min、8~12min等);然后再以8~12℃/min(例如9℃/min、10℃/min等)升温至190~210℃(例如200℃等),并保持0.5~2min(例如1min、1.5min等);后再以25~35℃/min(例如30℃/min等)最终升温至230~255℃(例如240℃、250℃等),并保持1~3min(例如1.5min、2min、2.5min等)。
[0173]
通过上述的温控方式,使得强极性物质能够更好的渗入进色谱柱固相结构中,进而能够将这些物质与弱极性的乙基麦芽酚分离。
[0174]
对于气质联用的其他操作条件,例如质谱的操作条件等,可以参照前文《第一方面》公开的内容进行。
[0175]
此外,本发明中的“安全标准”,如前所述,可以是地方、地区、国家、区域或国际组织指定的安全标准,例如这样的安全标准中容忍的对象产品中乙基麦芽酚的含量为不超过5μg/kg~25μg/kg,例如不超过10μg/kg、不超过15μg/kg或不超过20μg/kg等等。在本发明一些优选的实施方案中,遵照《食用植物油中乙基麦芽酚的测定(bjs 201708)》其食用油中的乙基麦芽酚的检出限为25μg/kg(即安全标准阈值浓度)。
[0176]
实施例
[0177]
以下,将通过实施例对本发明做出进一步的说明。
[0178]
测试仪器:
[0179]
气相色谱-质谱联用仪,美国赛默飞世尔科技,trace 1310-isq。
[0180]
冷冻离心机,sigma,2-16kl。
[0181]
石英两通,用于气相色谱柱连接,美国赛默飞,c-qsc10。
[0182]
原材料:
[0183]
浓香菜籽油、精炼四级菜籽油样品均来源于益海(广汉)粮油饲料有限公司。
[0184]
检测系统基准保留时间与丰度比的确定:
[0185]
1)标准样品:
[0186]
使用乙腈溶剂配制乙基麦芽酚的浓度为200μg/kg的标准样品。
[0187]
2)气质联用测试系统:
[0188]
气相色谱条件:
[0189]
进样方式:气相色谱仪进样口温度:250℃;不分流进样;进样量:1μl。采用tg-1701ms(规格:30m*0.25mm*0.25um)强极性色谱柱对乙基麦芽酚进行分离后测定;
[0190]
升温程序为:初始柱温60℃,保持4min,以3℃/min升温至100℃,保持10min,再以30℃/min升温至250℃,保持5min。
[0191]
色谱载气:高纯氦(纯度>99.999%),流速:1.0ml/min。
[0192]
质谱条件:
[0193]
选择离子扫描(sim)监测;电离方式:电子轰击电离源(ei);电离能量:70ev;传输线温度300℃;离子源温度300℃;溶剂延迟:10min。以保留时间、离子丰度比定性;定量离子为140离子,定性离子为:139、125、97离子。
[0194]
使用上述气质联用系统对标准样品进行测试确定保留时间以及基质谱离子的基准丰度比,参见图1a和图1b,其中气相色谱保留时间为28.91min。
[0195]
实施例1(基于强极性色谱柱的气质联用定量检测方法):
[0196]
本实施例中以强极性色谱柱分离目标物后,单四级杆质谱定量分析;包括以下步骤:
[0197]
a、加标样品配置
[0198]
采用待测油样(浓香菜籽油)稀释标准样品,配制标准加入法工作曲线用加标样品;所述待测油样为菜籽油,加标样品为用待测油样作为基质稀释剂,配制成浓度为15μg/kg、25μg/kg、50μg/kg、100μg/kg、200μg/kg加标样品;
[0199]
b、前处理
[0200]
混匀上述各个加标样品和待测油样后,各准确称取2g
±
0.01g于15ml玻璃离心管中,加入2ml色谱级乙腈,涡旋5min,将离心管样品放置于离心机中,以4000r/min的转速、4℃条件下,离心5min,用移液枪吸取收集最上层清液100μl于内插管中盖好瓶盖后,在气相色谱-质谱联用仪上待进样分析;
[0201]
c、气相色谱条件:
[0202]
进样方式:气相色谱仪进样口温度:250℃;不分流进样;进样量:1μl。采用tg-1701ms(规格:30m*0.25mm*0.25um)强极性色谱柱对乙基麦芽酚进行分离后测定;
[0203]
升温程序为:初始柱温60℃,保持4min,以3℃/min升温至100℃,保持10min,再以30℃/min升温至250℃,保持5min。
[0204]
色谱载气:高纯氦(纯度>99.999%),流速:1.0ml/min。
[0205]
d、质谱条件:
[0206]
选择离子扫描(sim)监测;电离方式:电子轰击电离源(ei);电离能量:70ev;传输线温度300℃;离子源温度300℃;溶剂延迟:10min.以保留时间、离子丰度比定性;定量离子为140离子,定性离子为:139、125、97离子。
[0207]
e、测试过程:
[0208]
在定量分析前,对待测样品经前处理得到的检测样品进行检测,以色谱出峰时间、质谱离子丰度比作为定性依据进行判断,经过与图1的保留时间和基准离子丰度对比,可以判断待测样品中含有乙基麦芽酚。
[0209]
将前处理好后的标准工作曲线试样溶液(加标样品溶液)、待测样品溶液前处理后得到的检测样品分别注入气相色谱-质谱联用仪中进行测试。
[0210]
以峰面积为纵坐标,标准加入含量(μg/kg)为横坐标,采用标准加入法绘制定量曲线后,以外标法对待测样品进行定量测定。得到线性方程分别为y=8.145
×
103x+1.992
×
104,相关系数为0.9984(参见图2)。
[0211]
同时采用待测样品配制不同浓度的标准加入试样,按上述流程进行验证,得到回收率及平行性数据;以考察方法的准确度、精密度。得到回收率为95-110%,rsd为8%。
[0212]
图3显示使用实施例1中的待测样品前处理后的检测样品的气相色谱图中(同时叠加了100μg/kg加标样品的色谱图),乙基麦芽酚的峰型无干扰,且定量检测结果显示待测油样的乙基麦芽酚含量为7μg/kg(10倍信噪比)。
[0213]
参考例1:
[0214]
将实施例1的气相色谱柱的升温程序替换为以下升温程序以外,其他条件与实施例1相同:
[0215]
初始柱温60℃,保持0min,以5℃/min升温至120℃,保持0min,再以35℃/min升温至250℃,保持5min。
[0216]
初步定量结果约100μg/kg,经质谱图分析,发现在此出峰的左右1/2峰宽处质谱图丰度比与标样不一致,m/z125离子已完全消失(原25%丰度),说明有共流出峰情况,峰未完全分开,有其他化合物干扰。
[0217]
实施例2
[0218]
使用实施例1的检测方法对工厂榨油车间精炼四级菜籽油样品的检测结果,图4为实际样品图谱与标准工作曲线点(25μg/kg标准加入点)叠加图谱,tg-1701ms色谱柱;说明精炼四级菜籽油为完全未出峰,无任何干扰,结果为0(不存在乙基麦芽酚)。
[0219]
另外,图5对图3与图4中各种样品的检测图谱叠加,这也反应了本发明测试方法的有效性和可靠性。
[0220]
需要说明的是,尽管以具体实例介绍了本发明的技术方案,但本领域技术人员能够理解,本公开应不限于此。
[0221]
以上已经描述了本公开的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。本文中所用术语的选择,旨在最好地解释各实施例的原理、实际应用或对市场中的技术的改进,或者使本技术领域的其它普通技术人员能理解本文披露的各实施例。
[0222]
产业上的可利用性
[0223]
本发明所述的气质联用的检测方法可以在工业上用于食用油中乙基麦芽酚及其含量的可靠检测。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1