一种米拉贝隆杂质的制备及分离检测方法与流程
1.本发明涉及药物化学领域,具体涉及一种米拉贝隆杂质的制备及分离检测方法。
背景技术:
2.米拉贝隆(mirabegron),化学名为(r)-2-(2-氨基噻唑-4-基)-4'-[2-[(2-羟基-2-苯基乙基)氨基]乙基]乙酸酰基苯胺,分子式为c
21h24
n4o2s,结构如下:
[0003][0004]
米拉贝隆是第一个用于治疗膀胱过度活动症的β3-肾上腺素受体激动剂类药物,使用这种药物可使逼尿肌平滑肌在膀胱充盈-排尿周期的存储期变得松弛,从而促进增加膀胱容量,主要用于治疗伴急迫性尿失禁、尿急、尿频症状的膀胱过度活动症(oab)。
[0005]
研究表明,在米拉贝隆原料药(api)和制剂样品放置稳定性实验中发现会产生一个接近定量限的未知杂质,3~6月稳定性样品检测含量高达0.07%~0.09%,该杂质可能对药品的安全存在隐患,给人体带来危害,因此需要对此杂质进行充分研究。
技术实现要素:
[0006]
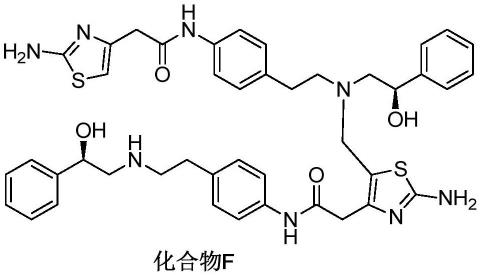
发明人经过研究,确认了米拉贝隆原料药和制剂样品放置稳定性实验中产生的杂质的结构,为下式所示的化合物f。为了更深入研究此杂质,需要能够简便地获得高质量的化合物,并且能够分离检测其含量。
[0007][0008]
为此,本发明第一方面提供一种制备化合物f的方法。一种制备化合物f的方法,包括:在dmf溶剂中,米拉贝隆与二卤代烷烃,加热至45℃-65℃反应,反应完毕,经后处理,制得化合物f。
[0009][0010]
在一些实施方式中,一种制备化合物f的方法,包括:在dmf溶剂中,米拉贝隆、多聚甲醛与酸在20℃-40℃反应,反应完毕,经后处理,制得化合物f。
[0011]
所述二卤代烷烃为氯代、溴代或者溴氯代烷烃化合物。在一些实施例中,所述二卤代烷烃为溴氯甲烷。
[0012]
所述二卤代烷烃与米拉贝隆的投料摩尔比可以为0.3:1-1.5:1。在一些实施例中,所述二卤代烷烃与米拉贝隆的投料摩尔比为0.6:1。在一些实施例中,所述二卤代烷烃与米拉贝隆的投料摩尔比为1:1。
[0013]
所述米拉贝隆与dmf溶剂的质量体积比可以为0.10g/ml-0.50g/ml。在一些实施例中,所述米拉贝隆与dmf溶剂的质量体积比为0.17g/ml。在一些实施例中,所述米拉贝隆与dmf溶剂的质量体积比为0.25g/ml。在一些实施例中,所述米拉贝隆与dmf溶剂的质量体积比为0.35g/ml。
[0014]
所述多聚甲醛与米拉贝隆的投料摩尔比可以为1:1-3:1。在一些实施例中,所述多聚甲醛与米拉贝隆的投料摩尔比为2:1。
[0015]
所述酸选自盐酸和乙酸中的至少一种。在一些实施例中,所述酸为乙酸。
[0016]
以体积计,所述酸不超过dmf溶剂体积的5%。在一些实施例中,所述酸为dmf溶剂体积的4.17%。
[0017]
所述方法中,反应时间为24h-36h。在一些实施例中,所述反应时间为30h。
[0018]
所述后处理包括:反应结束后,往反应液中加入氯化钠水溶液和2-甲基四氢呋喃,混匀后静置,分液,水相再用2-甲基四氢呋喃萃取一次,合并有机相,减压浓缩,干燥,再经液相制备,制得化合物f。在一些实施方式中,所述后处理包括:反应结束后,往反应液中加入10%氯化钠溶液和2-甲基四氢呋喃,充分混匀后静置,分液,水相再用2-甲基四氢呋喃萃取一次,合并有机相,于45℃减压浓缩,干燥,再经液相制备(色谱柱:kromasil c18,250mm
×
50mm,10μm;流动相a:0.1%氨水溶液,流动相b:乙腈;流速:100ml/min;检测波长:236nm;柱温:不控制;以流动相a:流动相b=53:47洗脱,每次进样0.5ml,收集出峰时间9.8min-10.8min的溶液),制得化合物f。
[0019]
本发明提供的方法,制备得到的化合物f的纯度不低于95%。
[0020]
在一些实施方式中,本发明提供的方法,得到的化合物f纯度大于95.0%或97.0%或98.0%或99.0%。
[0021]
本发明涉及到的反应,可以采用液相色谱法(hplc)监控反应终点;当检测表明式米拉贝隆(即(r)-2-(2-氨基噻唑-4-基)-4'-[2-[(2-羟基-2-苯基乙基)氨基]乙基]乙酸酰基苯胺)的峰面积≤5.0%时,可视为反应结束。
[0022]
本发明第二方面提供一种化合物f的分离检测方法。一种化合物f的分离检测方法,包括:以hplc检测样品溶液,检测时采用十八烷基键合硅胶(c18)为填料的色谱柱,流动
相分为a相和b相,a相为磷酸盐缓冲溶液,b相为乙腈。
[0023]
所述色谱柱的填料为超惰性ace硅胶颗粒。在一些实施例中,所述色谱柱为ace excel 3super c18。
[0024]
所述a相中的磷酸盐缓冲溶液选自磷酸钠缓冲液和磷酸钾缓冲液中的至少一种。在一些实施方式中,所述a相中的磷酸盐缓冲溶液为磷酸二氢钾缓冲液。
[0025]
所述磷酸盐缓冲溶液的浓度为0.01mol/l-0.05mol/l。在一些实施方式中,所述磷酸盐缓冲溶液的浓度为0.02mol/l。
[0026]
按照体积比计,所述洗脱程序为:
[0027]
时间(min)流动相a流动相b0851510.00752520.00307025.003070。
[0028]
在一些实施方式中,所述的化合物f的分离检测方法,可包括以下步骤或按以下步骤实现:
[0029]
1)取待测样品适量,加稀释剂配制成样品溶液;
[0030]
2)设置仪器参数:流动相的流速、色谱柱柱温、样品盘温度、洗脱程序、检测波长、后运行时间;
[0031]
3)取步骤1)的样品溶液,注入高效液相色谱仪,记录色谱图,完成化合物f的分离检测。
[0032]
步骤1)中,每1ml样品溶液中可含待测样品0.5mg-1mg。在一些实施例中,步骤1)中,每1ml样品溶液含待测样品0.6mg,0.7mg或0.8mg。
[0033]
步骤1)中,所述稀释剂为甲醇水的混合溶液(v:v=1:1)。
[0034]
步骤2)中,所述流动相的流速可为0.8ml/min-1.2ml/min。在一些实施例中,步骤2)中,流动相的流速为0.9ml/min。在一些实施例中,步骤2)中,流动相的流速为1.0ml/min。在一些实施例中,步骤2)中,流动相的流速为1.1ml/min。
[0035]
步骤2)中,所述样品溶液进样量为10μl-30μl。在一些实施例中,所述样品溶液进样量为15μl。在一些实施例中,所述样品溶液进样量为20μl。在一些实施例中,所述样品溶液进样量为25μl。
[0036]
步骤2)中,所述色谱柱柱温20℃-30℃。在一些实施方式中,所述色谱柱柱温20℃-25℃。在一些实施方式中,所述色谱柱柱温25℃-30℃。在一些实施方式中,所述色谱柱柱温23℃。在一些实施方式中,所述色谱柱柱温25℃。在一些实施方式中,所述色谱柱柱温28℃。
[0037]
步骤2)中,所述样品盘温度为4℃-10℃。在一些实施例中,所述样品盘温度为4℃。在一些实施例中,所述样品盘温度为5℃。在一些实施例中,所述样品盘温度为8℃。
[0038]
步骤2)中,所述检测波长为235nm-260nm。在一些实施方式中,所述检测波长为240nm。在一些实施方式中,所述检测波长为250nm。在一些实施方式中,所述检测波长为255nm。
[0039]
步骤2)中,所述后运行时间为6min。
[0040]
在一些实施例中,所述的分离检测化合物f的方法,包括以下步骤或可按以下步骤实现:
[0041]
1)取待测样品适量,加稀释剂配制成浓度为样品溶液,每1ml样品溶液中可含待测样品0.5mg-1mg;
[0042]
2)设置流动相流速0.8ml/min-1.2ml/min,色谱柱柱温20℃-30℃,样品盘温度为4℃-10℃,检测波长为235nm-260nm,洗脱程序为0min:流动相a:流动相b=85:15(v:v),10min:流动相a:流动相b=75:25(v:v),20min-25min:流动相a:流动相b=30:70(v:v);后运行6min;
[0043]
3)取步骤1)的样品溶液,注入高效液相色谱仪,记录色谱图,完成化合物f的分离检测。
[0044]
高效液相色谱仪可为美国agilent 1260型高效液相色谱系统及工作站或其他适宜可行的系统。
[0045]
在一些实施例中,所述的一种分离检测化合物f的方法,包括:以hplc检测样品溶液,检测时采用的色谱柱选自ace excel 3super c18;流动相分为a相和b相,a相为0.02mol/l磷酸二氢钾缓冲液,b相为乙腈;所述洗脱程序为0min:流动相a:流动相b=85:15(v:v),10min:流动相a:流动相b=75:25(v:v),20min-25min:流动相a:流动相b=30:70(v:v);后运行6min;流动相流速为0.8ml/min-1.2ml/min;色谱柱柱温20℃-30℃;检测波长为235nm-260nm。
[0046]
在一些实施例中,所述的一种分离检测米拉贝隆杂质的方法,包括:以hplc检测样品溶液,检测时采用的色谱柱选自ace excel 3super c18;流动相分为a相和b相,a相为0.02mol/l磷酸二氢钾缓冲液,b相为乙腈;所述洗脱程序为0min:流动相a:流动相b=85:15(v:v),10min:流动相a:流动相b=75:25(v:v),20min-25min:流动相a:流动相b=30:70(v:v);后运行6min;流动相流速为1.0ml/m in;色谱柱柱温25℃;检测波长为250nm。
[0047]
采用本发明所述的分离方法,分离检测米拉贝隆杂质化合物f的时间可在35分钟以内。
[0048]
采用本发明所述的分离检测方法,能够有效地、较好地分离检测化合物f,从而有效地保证药品的质量。
[0049]
术语说明:
[0050]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0051]
本发明中,如“化合物a”和“式a所示的化合物”的表述,表示的是同一个化合物。
[0052]
术语“室温”是指温度在大约20℃-35℃或大约23℃-28℃或大约25℃。
[0053]
在上文或下文的内容中,无论是否使用“大约”或“约”等字眼,所有在此公开了的数字均为近似值。每一个数字的数值有可能会出现1%、2%、5%、7%、8%或10%等差异。
[0054]
本发明的方法,可以采用液相色谱法(hplc)检测反应物纯度。
附图说明
[0055]
图1示实施例1中化合物f的质谱检测结果图;
[0056]
图2示实施例3中空白溶液的hplc图谱,横坐标表示时间(time),单位分钟(min),纵坐标表示电信号,单位(mau);
[0057]
图3示实施例3中对照品溶液的hplc图谱,横坐标表示时间(time),单位分钟(min),纵坐标表示电信号,单位(mau);
[0058]
图4示实施例3中供试品加标溶液1的hplc图谱,横坐标表示时间(time),单位分钟(min),纵坐标表示电信号,单位(mau);
[0059]
图5示实施例3中供试品加标溶液2的hplc图谱,横坐标表示时间(time),单位分钟(min),纵坐标表示电信号,单位(mau)。
具体实施方式
[0060]
为了使本领域的技术人员更好地理解本发明的技术方案,下面进一步披露一些非限制实施例,对本发明作进一步的详细说明。
[0061]
本发明所使用的试剂均可以从市场上购得或者可以通过本发明所描述的方法制备而得。
[0062]
本发明实施例中,所述的杂质含量计算公式如下:
[0063][0064]
式中:ws为杂质对照品溶液称样量,mg;
[0065]at
为供试品溶液中杂质的峰面积;
[0066]dt
为供试品溶液的稀释倍数;
[0067]as
为杂质对照品溶液(供试品溶液前后各1针)杂质峰面积的平均值;
[0068]ds
为杂质对照品溶液的稀释倍数;
[0069]wt
为供试品的称样量,mg。
[0070]
本发明中,hplc表示高效液相色谱法,ml表示毫升,μl表示微升,g表示克,nm表示纳米,mm表示毫米,mg表示毫克,min表示分钟,h表示小时,mg/ml表示毫克每毫升,℃表示摄氏度,dmf指二甲基甲酰胺。
[0071]
本发明中实施例中米拉贝隆杂质化合物1、杂质化合物2、杂质化合物3、杂质化合物4结构如下所示:
[0072][0073]
为了进一步理解本发明,下面结合实施例对本发明进行详细说明。
[0074]
实施例1:合成化合物f
[0075]
室温下向烧瓶中加入米拉贝隆(8.0g,1.0eq),溴氯甲烷(1.56g,0.6eq)和dmf(48ml),升温至55℃搅拌反应,搅拌30h后取样,hplc检测,反应完毕,往体系缓慢加入约10%氯化钠水溶液(290ml)和2-甲基四氢呋喃(120ml),搅拌30min后静置分液,水相再用2-甲基四氢呋喃(120ml)萃取一次,合并有机相,有机相于45℃减压浓缩除去溶剂,得到黄色油状物4.5g,纯度36.11%,再经液相制备纯化(色谱柱:kromasil c18,250mm
×
50mm,10μm;流动相a:0.1%氨水溶液,流动相b:乙腈;流速:100ml/min;检测波长:236nm;柱温:不控制;以流动相a:流动相b=53:47洗脱,每次进样0.5ml,收集出峰时间9.8min-10.8min的溶液),制得化合物f,黄色固体0.85g,纯度为95.81%,收率为10.5%。
[0076]
化合物f的质谱图如图1所示,esi-ms(m/z):805.3(m+h)
+
827.3(m+na)
+
;
[0077]1h nmr(400mhz,dmso-d6)δ:10.02(s,2h),7.50(dd,j=1.2hz,j=5.6hz,4h),7.28~7.34(brm,8h),7.23(m,2h),7.11(d,j=1.2hz,2h),7.08(d,j=1.2hz,2h),6.91(s,2h),6.78(s,2h),6.30(s,1h),5.26(brs,1h),4.84(s,1h),4.67(m,1h),4.62(m,1h),3.76(m,2h),3.47(d,4h),2.61
–
2.80(m,12h),2.19(s,1h);
[0078]
13
c nmr(150mhz,dmso-d6)δ:168.71,168.35,168.27,167.04,151.95,146.35,145.03,144.97,142.91,139.67,137.69,137.59,135.54,135.45,129.28,129.23,128.39,128.33,127.23,126.59,126.34,125.39,119.47,119.43,119.41,103.05,71.88,71.27,
61.83,57.94,55.72,51.20,49.83,40.26,37.83,35.77,32.51。
[0079]
实施例2:合成化合物f
[0080]
室温下烧瓶中加入米拉贝隆(4.0g,1eq),多聚甲醛(0.6g,2.0eq)、乙酸(1ml)和dmf(24ml),升温至30℃搅拌反应,搅拌30h后取样,hplc检测,反应完毕,往体系缓慢加入约10%氯化钠水溶液(290ml)和2-甲基四氢呋喃(120ml),搅拌30min后静置分液,水相再用2-甲基四氢呋喃(120ml)萃取一次,合并有机相,有机相于45℃减压浓缩除去溶剂,得到黄色油状物2.3g,纯度40.23%,再经液相制备纯化(色谱柱:kromasil c18,250mm
×
50mm,10μm;流动相a:0.1%氨水溶液,流动相b:乙腈;流速:100ml/min;检测波长:236nm;柱温:不控制;以流动相a:流动相b=53:47洗脱,每次进样0.5ml,收集出峰时间9.8min-10.8min的溶液),制得化合物f:黄色固体0.45g,纯度为96.23%,收率为11.1%。
[0081]
实施例3:专属性考察
[0082]
仪器与条件:
[0083][0084]
实验步骤:
[0085]
流动相a相:取磷酸二氢钾2.72g,加水1000ml使溶解,用氢氧化钾试液调ph值至5.6,用0.2μm水系滤膜过滤,超声10min,即得;
[0086]
流动相b相:乙腈;
[0087]
稀释剂/空白溶液:甲醇-水(1:1,v/v)的混合溶液;
[0088]
对照品储备液:取化合物f对照品6mg,置100ml容量瓶中,加甲醇超声使溶解并稀释至刻度,摇匀,即得;
[0089]
对照品溶液:精密移取对照品储备液1ml至100ml容量瓶中,加稀释剂至刻度,摇匀,即得;
[0090]
杂质对照品溶液:取米拉贝隆杂质化合物1、杂质化合物2、杂质化合物3、杂质化合物4对照品各9mg至100ml容量瓶中,加甲醇超声使溶解并稀释至刻度,摇匀;分别精密移取上述各溶液1ml、对照品储备液1ml至100ml容量瓶中,加稀释剂至刻度,摇匀,即得;
[0091]
供试品溶液:取供试品约30mg,精密称定,置50ml容量瓶中,加稀释剂超声溶解并稀释至刻度,摇匀,即得;
[0092]
供试品加标溶液1:取供试品约30mg,精密称定,置50ml量瓶中,加对照品溶液超声使溶解并稀释至刻度,摇匀,即得;
[0093]
供试品加标溶液2:取供试品约30mg,精密称定,置50ml量瓶中,加杂质对照品溶液超声使溶解并稀释至刻度,摇匀,即得。
[0094]
待系统平衡后,取空白溶液、对照品溶液、供试品溶液、供试品加标溶液1、供试品加标溶液2按照以下的序列表(表a)及上述条件进行hplc分析,记录色谱图,如图2-图5所
示,结果见表1。
[0095]
表a
[0096]
样品名进样针数空白溶液1~2对照品溶液3供试品溶液1供试品加标溶液11供试品加标溶液21对照溶液1
[0097]
结果分析:
[0098]
表1系统适用性结果
[0099][0100][0101]
实施例4:定量限(loq)和检测限(lod)考察
[0102]
仪器与条件:
[0103][0104]
实验步骤:
[0105]
流动相a相:取磷酸二氢钾2.72g,加水1000ml使溶解,用氢氧化钾试液调ph值至5.6,用0.2μm水系滤膜过滤,超声10min,即得;
[0106]
流动相b相:乙腈;
[0107]
稀释剂/空白溶液:甲醇-水(1:1,v/v)的混合溶液;
[0108]
对照品储备液:取化合物f对照品6mg,置100ml容量瓶中,加甲醇超声使溶解并稀释至刻度,摇匀,即得;
[0109]
对照品溶液:精密移取对照品储备液1ml至100ml容量瓶中,加稀释剂至刻度,摇匀,即得;
[0110]
定量限(loq)溶液:精密移取对照品溶液5ml,置10ml容量瓶中,加稀释剂稀释至刻度,摇匀,即得(0.3μg/ml);
[0111]
检测限(lod)溶液:精密移取对照品溶液2ml,置10ml容量瓶中,加稀释剂稀释至刻度,摇匀,即得(0.12μg/ml)。
[0112]
待系统平衡后,取空白溶液、定量限溶液与检测限溶液进样,按照以下的序列表(表b)及上述条件进行hplc分析,记录色谱图,结果见表2。
[0113]
表b
[0114]
样品名进样针数空白溶液1~2检测限溶液3定量限溶液3
[0115]
表2
[0116][0117][0118]
实施例5:样品检测
[0119]
仪器与条件:
[0120][0121]
实验步骤:
[0122]
流动相a相:取磷酸二氢钾2.72g,加水1000ml使溶解,用氢氧化钾试液调ph值至5.6,用0.2μm水系滤膜过滤,超声10min,即得;
[0123]
流动相b相:乙腈;
[0124]
稀释剂/空白溶液:甲醇-水(1:1,v/v)的混合溶液;
[0125]
对照品储备液:取化合物f对照品6mg,置100ml容量瓶中,加甲醇超声使溶解并稀释至刻度,摇匀,即得;
[0126]
对照品溶液:精密移取对照品储备液1ml至100ml容量瓶中,加稀释剂至刻度,摇匀,即得;
[0127]
杂质对照品溶液:取米拉贝隆杂质化合物1、杂质化合物2、杂质化合物3、杂质化合物4对照品各9mg至100ml容量瓶中,加甲醇超声使溶解并稀释至刻度,摇匀;分别精密移取上述各溶液1ml、对照品储备液1ml至100ml容量瓶中,加稀释剂至刻度,摇匀,即得;
[0128]
供试品溶液:取待测样品3份,每份样品称取约30mg,精密称定,置50ml容量瓶中,加稀释剂超声溶解并稀释至刻度,摇匀,即得;其中,api指米拉贝隆原料药;api加速3月指将原料药按照中国药典稳定性条件在温度40℃
±
2℃,湿度75%
±
5%放置3月;制剂加速6月指将制剂样品按照中国药典稳定性条件在温度40℃
±
2℃,湿度75%
±
5%放置6月。
[0129]
待系统平衡后,取空白溶液、对照品溶液、供试品溶液按照以下的序列表(表c)及上述条件进行hplc分析,记录色谱图,结果见表3。
[0130]
表c
[0131][0132][0133]
表3
[0134][0135]
本发明的方法已经通过较佳实施例进行了描述,相关人员明显能在本发明内容、精神和范围内对本文所述的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1