评估生物样品中与分子效应相关的分子变化的方法与流程
本发明涉及一种手动或自动评估生物样品内跟随至少一种感兴趣分子的浓度或强度的分子变化的方法。更具体来说,本发明涉及一种方法,其中将成像技术用于检测生物样品内与感兴趣分子(例如活性化合物)的存在相关联的相对于分子生物标志物的变化。本发明允许定义与活性化合物位置有关的区域,以便计算跟随感兴趣分子的浓度的分子变化的评分、比率或任何值。
本发明的方法可应用于涉及研究生物样品中感兴趣分子的行为的所有领域。本发明的方法可有利地用于蛋白质组学、多肽组学、脂质组学、代谢组学、糖组学或制药学研究,以便筛选候选分子并评估它们的治疗或诊断潜力。
背景技术:
在制药、保健品、农用化学或化妆品行业中,活性化合物的开发需要在临床前早期阶段在体外细胞模型中或晚些时候在体内动物模型中鉴定作用机制(moa)、功效和毒性。在晚些时候,将这些测定法发展并转移到临床阶段中,以验证药物在人类中的功效和毒性。质谱(ms)、免疫组织化学(ihc)、酶联免疫吸附测定法(elisa)、原位杂交(ish)、聚合酶链反应(pcr)是在不同阶段用于分子变化评估和定量的黄金标准方法。所有这些方法都被开发用于定量样品中的分子并鉴定它们在药物功效或毒性中的作用。
所有这些方法通常被用于通过评估和量化诊断、预后、功效或对治疗的反应的生物标志物来研究组织反应。当用于对照和用药(使用活性化合物)样品时,质谱是最先进的方法之一,因为它不需要标记待定量的感兴趣分子。质谱将关于所述活性化合物的生物学影响的分子信息提供为生命系统中脂质、代谢物、肽或蛋白质浓度的提高或降低。其他技术(例如if、ihc、elisa、ish、分光光度法或uv荧光)允许评估和定量使用标记的抗体或与靶结合的核酸序列所靶向的肽、蛋白质、rna(核糖核酸)和dna(脱氧核糖核酸)。
在制药工业中,药效学是科学家研究在给药后的某个时间药物的影响,并评估靶分子(生物标志物)变化或非靶分子变化(在更大规模上研究药物反应并鉴定潜在的未知化合物)的领域。
在组织水平上,迄今为止描述的所有使用的方法都将组织作为一个整体进行比较,这意味着丢失了大量信息。这些实际方法使用整个组织或组织切片中分子变化的相对或绝对定量,并且没有考虑所述相关活性化合物的浓度在组织、细胞或其他特定感兴趣区域中所处的位置。所述方法没有考虑药物的定位对其邻域的影响。
在细胞水平上,当前的基于细胞活力的测量由于对所有细胞进行分析而没有与渗透或进入细胞的药物或活性化合物的相对或绝对浓度关联,因此通常会导致有偏差的响应估计。
因此,对于允许以更高的准确度和精确度将活性化合物的浓度与功效的生物标志物关联,以便研究细胞对活性化合物的反应的方法,存在着需求。
技术实现要素:
在这种情况下,本发明人现在提出了一种方法,所述方法将感兴趣化合物(例如药物候选物)在靶组织中的鉴定和定位与所述感兴趣化合物所在区域中的分子变化的图像分析相结合。观察所述感兴趣化合物的位置并选择它具体所在的区域和/或它呈现出不同浓度的区域,允许在非常精细的尺度上研究对所述化合物的响应和相关生物标志物的变化。根据本发明,不同样品之间或同一样品的不同区域之间的比较导致对可能与所述感兴趣化合物的存在和/或浓度相关或不相关的分子环境的了解。本发明的方法还允许研究感兴趣化合物的分子影响,并通过比较所述感兴趣化合物与相关生物标志物(例如功效或毒性的标志物)之间不同的计算比率对其进行评分,导致了解所述感兴趣化合物在邻域水平上的影响(例如功效或毒性)。
因此,本发明的一个目的是提供一种对用药生物样品中至少一种感兴趣分子对至少一种分子标志物的影响进行离体或体外评估的方法,所述方法包括:
-选择用药生物样品,其以前已被暴露到所述至少一种感兴趣分子;
-使用分子成像方法检测所述用药生物样品中所述至少一种感兴趣分子的存在,并为所述至少一种感兴趣分子获得所述用药生物样品的分子图谱;
-根据光谱信息对所述用药生物样品的分子图谱进行空间分割,以获得所述用药生物样品中所述至少一种感兴趣分子的分割图谱;
-从所述分割图谱选择第一感兴趣区域(roi),所述第一roi具有所述至少一种感兴趣分子的第一强度;
-测量所述第一roi中至少一种分子标志物的强度或量;
-将所述第一roi中至少一种分子标志物的强度或量与第二roi中至少一种分子标志物的强度或量进行比较,其中从所述用药生物样品或另一个生物样品选择所述第二roi。
所述方法可以使用可以用分子成像方法分析的任何种类的生物样品来进行,包括组织样品、类器官、生物流体样品例如尿液样品、血浆样品、脑脊液以及细胞悬液。
在特定实施方式中,所述用药生物样品是通过预先对以前给药过所述感兴趣分子的动物进行采样而获得的组织切片。可选地或此外,所述用药生物样品已在体外与所述感兴趣分子接触过。
在特定实施方式中,所述感兴趣分子是候选分子,并且所述用药生物样品预先通过在以前给药过所述候选分子的动物中采样而获得。
在特定实施方式中,从所述用药生物样品的分割图谱选择所述第二roi,所述第二roi与所述第一roi在物理上不同,并具有所述感兴趣分子的第二强度。在另一个实施方式中,从第二生物样品选择所述第二roi,所述第二生物样品以前已暴露到剂量浓度与用于所述用药生物样品的剂量浓度不同的所述感兴趣分子,所述第二生物样品来自于与所述用药生物样品相同的生物学起源。可选地,从以前未暴露到所述感兴趣分子的第二生物样品选择所述第二roi,所述第二生物样品来自于与所述用药生物样品相同的生物学起源。在另一个实施方式中,从以前已暴露到第二感兴趣分子的第二生物样品选择所述第二roi,所述第二生物样品来自于与所述用药生物样品相同的生物学起源。
在特定实施方式中,第一与第二roi之间的比较允许鉴定至少一种对所述感兴趣分子的生物效应和反应特异的生物标志物。
在特定实施方式中,所述第一和第二roi已与对应于两种不同候选分子的两种感兴趣分子接触,并且所述第一roi与第二roi之间的比较允许区分所述候选分子。在这样的实施方式中,在所述第一和第二roi中比较的生物标志物可以相同或不同。
在另一个实施方式中,所述第一和第二roi已与几种(即超过一种)感兴趣分子接触,在所述roi之间所述感兴趣分子可以相同或不同。在特定实施方式中,两个roi均已与相同的两种或更多种感兴趣分子接触。
在特定实施方式中,在所述第一和第二roi中比较的生物标志物是相同的,即使所述第二roi的感兴趣分子不同于所述第一roi的感兴趣分子。因此,本发明的方法可用于评估和比较至少两种不同的感兴趣分子对至少一种分子标志物的影响。
本发明的方法可以利用选自mri成像、pet成像、ct成像、if、ish、ihc和质谱或细胞仪成像的分子成像方法来进行。本发明的方法特别适合于质谱成像,例如maldi、desi、lesa、la-icp-ms和sims,用于检测所述生物样品中所述感兴趣分子的存在,并由此在所述检测的基础上对所述生物样品作图。
在所选的roi中生物标志物的量或强度的测量可以使用分子成像方法或通过其他生物分析技术(hplc、lc-ms/ms、gc/ms、磁珠多重免疫测定法、elisa)来进行。
在特定实施方式中,所述方法包括由在一种或不同的感兴趣分子与分子标志物之间建立剂量效应曲线构成的步骤,其中roi中一种或多种感兴趣分子中的每一者的强度或量根据同一roi中所述分子标志物的强度或量来表示。
在特定实施方式中,通过激光捕获显微切割从所述生物样品提取所述roi。然后可以在进行所述生物分析法之前在培养基中培养所述roi,以例如通过比较所述第一与第二roi之间基因或转录本表达、脂质组学、多肽组学、蛋白质组学和/或代谢组学的变化来评估所述感兴趣分子的影响。
在特定实施方式中,所述感兴趣分子是治疗性抗体,并且其中所述治疗性抗体的存在通过将所述生物样品与标记的抗治疗性抗体的抗体相接触来检测。
附图说明
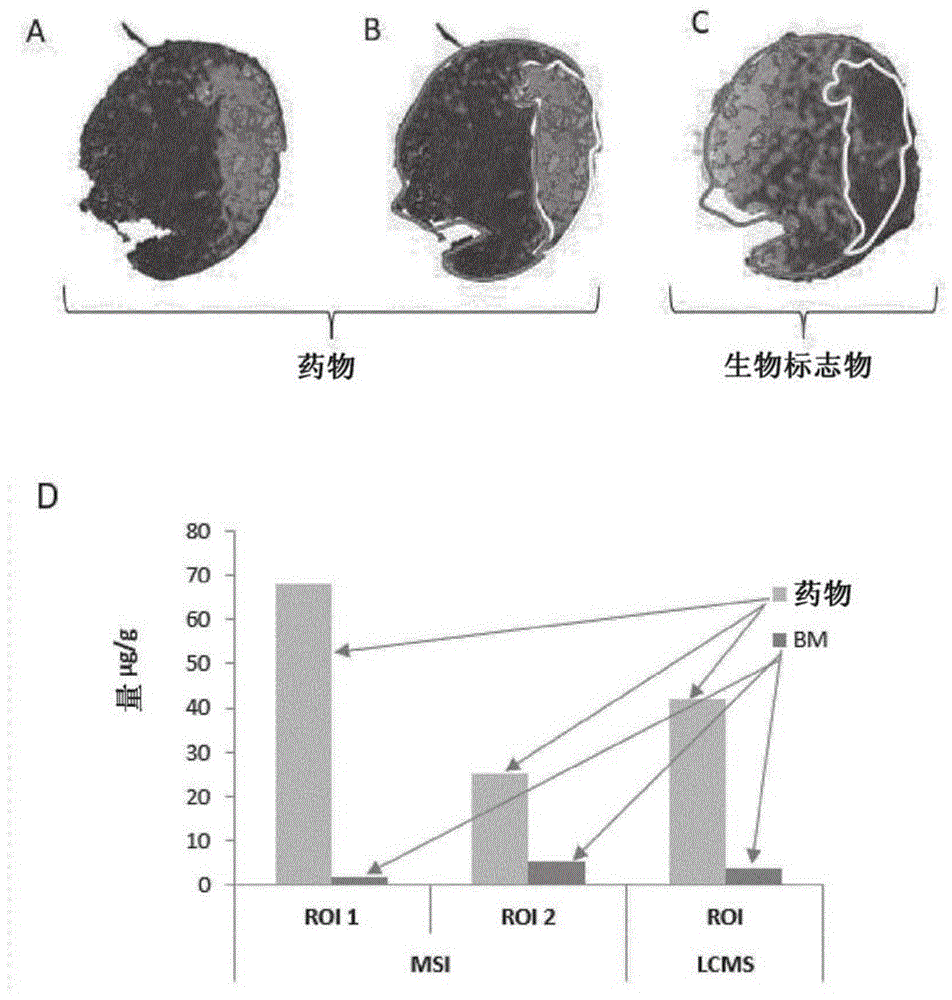
图1:已暴露于药物的用药生物样品的分子图谱(图1a)和将所述分子图谱分割成其中所述药物具有低强度(图1b中左侧)和其中所述药物具有高强度(图1b中右侧)的两个roi。所述分割允许区分受到所述药物不同影响的区域。所述两个roi显示出与所述药物相关的生物标志物的不同浓度(图1c),其可能与所述药物的浓度差异相关联,这与全局方法例如lcms相反(图1d)。
图2:未暴露到所述药物的组织样品的分子图谱(图2a–ctrl:对照)和已暴露到所述药物的组织样品的分子图谱(图2b–trt:治疗过的)。整个第一分子图谱被用作roi,而第二分子图谱被分割以选择具有高浓度的所述药物的roi。两个组织样品具有相同的起源(肿瘤组织)。来自于对照的roi显示出与所述药物的不存在相关的高浓度的生物标志物,而来自于治疗过的样品的roi显示出低浓度的所述生物标志物和高浓度的所述药物(图2c)。
图3:未暴露到所述药物的组织样品的分子图谱和来自于三份生物平行样的组织样品的分子图谱(图3a–ctrl、trt肿瘤1、trt肿瘤2和trt肿瘤3)。整个分子图谱被用作相应样品的roi。所有组织样品均具有相同起源(肿瘤组织)。来自于对照的roi显示出与所述药物的不存在相关的高浓度的生物标志物,而来自于治疗过的样品的roi显示出较低浓度的所述生物标志物,其可能与它们相应的药物浓度相关联(图3b)。
图4:已暴露到第一药物的组织样品的分子图谱(图4a–药物a)、已暴露到第二药物的组织样品的分子图谱(图4b–药物b)和相同的生物标志物的分子图谱(生物标志物,图4a和4b)。通过参考预先相对于所述药物的浓度鉴定的两个或三个roi中所述生物标志物的浓度来评估药物a和药物b的功效(图4c和4d)。
图5:高水平图像分割允许获得多区段图谱(像素图)。每个像素含有以μg/g组织为单位的从c1至c11浓度的药物(药物a或b)和生物标志物相关信息(图5a)。由此获得每个区段(像素)的药物生物效应,其对应于在所述药物的不同浓度或强度水平的基础上所述生物标志物的不同浓度或强度水平(图5b)。然后可以绘制不同曲线,其给出在单个组织中的药物剂量效应。ed50:有效剂量中值,即实现50%的所需反应所需的剂量。
图6:已暴露到药物的组织样品的分子图谱(图6a)、所述分子图谱的自动分割和roi的鉴定(图6b)。通过激光捕获显微切割进行roi提取的示意图(图6c)。
图7:epacadostat(epa)校准曲线/qmsi(a),epa校准曲线/lc-msms(b),通过qmsi和lc-ms/ms进行的epa定量(c)。
图8:epacadostat药物检测、定量和组织学定位。在1,5-dan基质沉积后,通过maldimsi对10μm厚的ct26对照和治疗过的肿瘤切片进行一式两份分析。epacadostat药物组织学定位显示出在所述治疗过的肿瘤中存在,但在对照肿瘤中不存在。对两份平行样(1和2)进行以μg/g为单位的epa绝对定量并示出在底部的表中(a)。在治疗过的ct26肿瘤中比较lc-ms/ms和qmsiepa定量,显示出两种技术之间17%的变化度(b)。示出了所有切片的标尺颜色和标尺条。
图9:靶暴露分析。选择显示出不同epa量的三个感兴趣区域(1、2和3)。roi1和2占整个肿瘤表面的38%和62%。发现了具体的epa定位,其显示61%集中在38%的表面中,并且39%存在于62%的剩余肿瘤表面上(a)。对连续组织切片进行半定量ido1免疫染色,并且插图1和2示出了在两个区域上ido1酶的不同表达水平(b)。所有这些叠加均使用imabiotech的多模式平台multimaging软件来进行。
图10:组织学定位和绝对定量。示出了在对照和治疗过的ct26肿瘤切片两者上的epa、trp和kyn组织学定位(a)。然后进行trp和kyn的qmsi和lc-ms/ms分析,然后从对照和治疗过的ct26肿瘤获得kyn/trp比率。使用qmsi和lc-ms/ms两种分析均注意到kyn/trp比率水平降低6倍(b)。
图11:归一化的trp和kyn校准曲线:x=以mg/g为单位的浓度;y=强度(图11a)。血浆和血液样品的kyn/trp比率(图11b)。
图12:暴露到反应和反应功效分析。根据在三个分割的roi中kyn的量,突出了epa的区域性药理作用和反应功效;其中15%的kyn集中在整个肿瘤的38%中(roi1),并且85%集中在roi2中(a)。也从分子图像提取了epa、kyn和乳酸的相对强度(b),以便显示出当epa高度或低度存在时的反应功效(c)。区域分割、相对和绝对定量使用imabiotech的多模式平台multimaging软件来进行。
发明详述
本发明的方法是基于在感兴趣分子的浓度的基础上对生物样品进行分割,并在同一生物样品的不同部分/区段中或在可能包含不同量的所述感兴趣分子和/或不同的感兴趣分子的不同生物样品之间比较与所述感兴趣分子的存在相关的分子变化。更具体来说,根据本发明,使用成像技术来检测和定位以前暴露到感兴趣化合物的生物样品中的所述化合物。然后在所述样品中所述化合物的强度的基础上对所述生物样品进行分割,并通过比较所述样品区段来分析分子变化。所述方法允许鉴定由于所述靶分子的存在和/或由于所述靶分子的不同剂量而增加或减少的分子(即生物标志物),甚至是在细胞水平上。本发明的方法还可以允许比较同一生物样品的两个或更多个区域之间感兴趣的化合物的分子影响,以便准确确定所述化合物在靶生物样品中的治疗和/或预防和/或毒性影响。然后可以鉴定新的潜在药物以及它们的剂量效应和最终相关的生物标志物。
根据本发明,使用成像技术来定位所述感兴趣分子,然后在整个所述生物样品中感兴趣分子的相对或绝对量的基础上使用图像分割来分析所述生物样品的分子图像。所选区段的分析允许确定可能归因于所述感兴趣分子的存在或特定剂量的分子变化,并由此选择所述感兴趣分子的有价值的生物标志物和/或筛选有价值的药物等。
与现有技术的方法相反,本发明的方法可以提供生物样品的不同部分中、包括同一生物组织的不同细胞之间与感兴趣分子相关的分子功效信息。因此可以评估靶生物样品中分子的实时影响。假定所述生物样品的一个分子图谱代表了所述化合物的一定浓度范围,可以计算每个图像像素的活性化合物/生物标志物比率,这将给出单一组织中的药物剂量效应。
生物样品的制备
根据本发明的方法,可以对以前已暴露到感兴趣分子的所有种类的生物样品进行分析。更通常来说,所述生物样品涵盖了表现出细胞活性的所有体系。这包括例如细胞培养物、3d体外模型例如球状体或微型器官、体内系统如动物模型、植物、异种移植组织、肿瘤和动物活检样品、生物流体等。在有利情况下,所述生物样品选自组织样品(例如活检样品)、生物流体样品(例如作为尿液样品、血浆样品、脑脊液)和细胞悬液。
在本发明的情形中,术语“组织”是指一组有功能的分组细胞。所述靶组织可以是一组具有相同起源的相似或不同的细胞、器官、器官的一部分、器官的任选地具有多细胞集合体的特定区域。例如,所述靶组织可以是位于器官内的肿瘤。在另一个实施方式中,所述组织样品是植物组织样品。更通常地,组织样品是指采取可以通过成像方法分析的形式的生物起源的任何类型的组织。
生物流体涵盖了来自于生物起源的所有流体,其包含细胞和/或从动物或植物获得。
在本发明的情形中,“暴露到”感兴趣分子意味着所述生物样品已与所述分子接触过。在本发明的情形中,已暴露到所述靶分子的生物样品被称为“用药或治疗过的生物样品”。相反,“对照样品”是指未暴露到所述靶分子的生物样品。根据本发明,对照样品与它所比较的用药样品具有相同的生物起源。例如,如果所述用药组织样品由在已暴露到所述靶分子的小鼠中取样的肝组织切片构成,则所述对照样品将由在未暴露到所述靶分子的小鼠中取样的肝组织切片构成。
在特定实施方式中,所述感兴趣分子以前已被给药到动物模型,优选为哺乳动物,包括人类和非人类哺乳动物,并且所述动物在所述方法的实施方式之前已被采样。在特定实施方式中,所述动物模型是非人类哺乳动物。在另一个特定实施方式中,所述动物模型是人类。
取决于所需研究,所述动物模型可以改变。专业技术人员了解哪种动物模型良好地适应于所评估的靶组织、感兴趣分子、生物学性质等。例如,在需要处死动物的候选分子的临床前试验的情况下,优选地使用非人类哺乳动物例如啮齿动物(小鼠、大鼠、兔、仓鼠等)。可以使用其他非人类哺乳动物,特别是猴、狗等。在其中生物样品可以在对动物没有有害副作用的情况下采样的某些情况下,可以使用人类作为动物模型(例如用于1-4期临床试验)。还可以使用其他动物模型例如鱼类、昆虫,以例如研究分子对环境或特定生态介质的影响。
根据本发明并且广义来说,可以使用所述靶分子的所有给药途径,例如肠内途径(即药物通过胃肠道的消化过程给药)或肠胃外途径(即胃肠道以外的其他给药途径)。例如,所述分子可以通过不同途径给药,例如表皮、硬膜外、动脉内、静脉内、皮下(具有特定定位)、心内、海绵体内注射、脑内、真皮内、肌肉内、骨内输注、腹膜内、鞘内、膀胱内、玻璃体内、鼻、口、直肠、阴道内或局部施用。
所述给药途径可以根据感兴趣分子、方法所靶向的组织等来选择。本发明的方法也可以允许选择最适合的给药途径。事实上,本发明的方法允许评估感兴趣分子跨过生物屏障到达所述靶组织的能力。
根据本发明,所述方法优选离体和/或体外进行。在某些情况下,也可以对活的完整动物进行体内分析。
在特定实施方式中,本发明的方法是离体分析,例如对组织切片的离体分析。在这种情况下,所述组织样品在给药后的给定时间(t1)采样。所述采样可以是活检,特别是当所述动物是人类时。根据一个实施方式,在将所述感兴趣分子给药到非人类动物后,将所述非人类动物处死并对感兴趣的组织样品进行采样。
在特定实施方式中,所述组织样品从新鲜组织、冷冻组织或例如用石蜡固定/包埋的组织获得。适合于获得例如几微米厚的薄组织切片的所有手段均可以使用。
如有需要,所述组织切片可以接受预处理,这特别地取决于待检测的分子、分析技术等。因此,可以对组织切片使用化学或生物化学试剂,以优化所述感兴趣分子和生物标志物的检测。例如,可以使用溶剂和/或去污剂,以允许检测限定类别的分子或改进分子从组织的直接提取。此外,可以使用能够切割肽或蛋白质的特异性酶,以便靶向例如在组织上具有与所述母体分子相同的定位和/或量的消化片段。还可以在组织上或组织中添加某些分析物,用于所述感兴趣分子的化学衍生,因为它解决了某些灵敏度问题,允许提高组织上的分布和定量研究。还可以在组织切片上进行抗体标记(偶联或不带有标签)或使用荧光标记分子或放射活性,以允许检测所述感兴趣分子和生物标志物。
也可以改变所使用的动物模型和/或所述靶组织和/或组织切片,以便修改它们结合或并入所述感兴趣分子的能力。因此,这种处理可以包括动物模型和/或靶组织和/或组织切片的化学或生物学修饰,其允许提高或抑制感兴趣分子对给定靶组织的渗透或靶向能力。这种处理可以在所述感兴趣分子给药之前、之后或同时进行。例如,在分子必须跨过血脑屏障(bbb)的情况下,在所述屏障中存在一些外排转运蛋白,其能够排出跨过bbb的所述分子。这些转运蛋白的作用可以使用抑制剂或对所述转运蛋白的基因或基因表达的遗传修饰例如“敲除”来调节(降低或抑制)。
在液体样品的情况下,可以将它在表面上干燥,以便产生所述干燥样品的ms图像,然后用msi来表征该样品。
如果使用需要基质的质谱成像、尤其是maldi或me-sims(基质增强二次离子质谱)来研究生物样品,则在有利情况下所述基质适应于所述感兴趣分子。例如,选择可以将覆盖的质量范围考虑在内。专业技术人员知道在现有的液体或固体基质中,根据所研究的分子和/或靶组织可以使用哪一种。同样地,基质的所有沉积方法均可以使用,特别是手动喷涂、自动喷涂、升华、过筛和微点样。
在另一个实施方式中,将所述感兴趣分子在体外或离体与生物样品例如组织样品、生物流体、细胞悬液等相接触。例如,所述生物样品是细胞悬液,将所述感兴趣分子添加到所述悬液,然后进行分析。可选地,可以将所述生物样品沉积在支持物上(并且在流体样品的情况下,可以任选地将它干燥),并将所述感兴趣分子喷涂在所述样品上,然后进行分析。
感兴趣分子的检测
对所述生物样品进行分析,以便检测所述生物样品中所述感兴趣分子的存在和任选的浓度。
这个步骤可以使用允许在体内、体外或离体准确地鉴定生物样品中的分子和将所述分子可视化的任何技术来进行。
尤其是,在体内分析的情况下,可以使用断层扫描技术例如磁共振成像(mri)、放射自显影、正电子发射断层扫描(pet)、单光子发射断层扫描、ct成像等。
在体外/离体分析的情况下,可以使用if、ihc、ish和质谱成像(msi)。可以使用诸如maldi成像(基质辅助激光解吸/电离)、ldi(激光解吸/电离)、desi(通过电喷雾解吸)、lesa(液体萃取表面分析)、laesi(激光烧蚀电喷雾电离)、dart(实时直接分析)、sims(二次离子质谱)、jedi(射流解吸电喷雾电离)的技术,并任选地与不同种类的质量分析仪如tof(飞行时间)、orbitrap、fticr(傅立叶变换离子回旋共振)、四级杆(单重或三重)等相组合。优选地,所述质谱成像选自maldi、desi、icp-ms和sims。
将实验参数例如质量范围、激光能量密度、激光聚焦等固定,以优化靶检测的强度、灵敏度和分辨率。因此,进行质谱图的获取以获得信号。从所述质谱图可以获取对靶分子研究有用的数据。对于数据处理来说,可以使用不同的光谱特征,例如质谱图上的峰强度、信噪比(s/n)、峰面积等。
作为一般规则,可以使用允许将生物样品内的分子可视化的所有技术。
感兴趣区域(roi)的选择
所述生物样品的分析至少为所述感兴趣分子提供了所述生物样品的分子图谱。也就是说,获得了整个所述生物样品中所述感兴趣分子的图像。所述图像允许直接将所述感兴趣分子在所述生物样品中的分布和浓度可视化。因此可以将所述生物样品的不同区域内所述分子的存在和/或不存在可视化,并且假设所述生物样品的一个分子图谱代表了浓度范围,也可以将所述不同区域之间的浓度差异可视化。然后将所述生物样品的分子图谱分割成不同区域。
所述生物样品的分割可以产生从像素到区段(包含多个像素)的各种不同尺寸的区域。所述分割可以手动或自动进行,以便获得以所述感兴趣分子的存在/不存在/强度/量(即所述感兴趣分子的光谱信息)为特征的不同区域。例如,所述分割可以使用选自基于区域的分割、边缘检测分割、基于簇集的分割、基于cnn中的弱监督学习的分割等的图像分割技术自动进行。然后可以选择感兴趣区域(roi)。
所述roi的尺寸可以彼此不同。roi可以是许多图像像素或区段的平均值。每个图像像素或区段也可以代表具有感兴趣分子的不同强度或浓度的roi。
在特定实施方式中,所述生物样品是已通过msi进行分析的组织切片。获得的图像显示了所述感兴趣分子的叠加分布。因此,在所述感兴趣分子的峰强度、峰面积或信噪比的基础上进行所述分割。
例如,所述感兴趣分子是候选分子,并且所述用药生物样品预先通过在以前给药过所述候选分子的动物中采样而获得。
在一个实施方式中,从所述用药生物样品的分割图谱选择所述第二roi,所述第二roi与所述第一roi在物理上不同,并具有所述感兴趣分子的第二强度。在有利情况下,所选的第一roi表现出最重要的信号(以及因此最重要的浓度),并且所选的第二roi不表现出所述感兴趣分子的任何信号(即被剥夺所述感兴趣分子)。可选地,所述用药生物样品的第二roi表现出所述感兴趣分子的低信号(以及因此低浓度)。
图1示出了使用msi为药物获得的组织切片的分子图谱。所述组织切片在以前给药过所述药物的动物中采样。所述分子图谱(图1a)是基于与所述分子相关的强度。从在所述组织切片上可视化的光谱数据,可以在同一组织切片内划出两个roi(图1b)。所述第一roi对应于所述组织样品的表现出所述分子的高信号的区域,而所述第二roi对应于所述组织样品的表现出所述分子的低信号的区域。
在另一个实施方式中,从以前未暴露到所述感兴趣分子的第二生物样品选择所述第二roi,所述第二生物样品来自于与所述用药生物样品(即对照样品)相同的起源。
在本发明的情形中,“相同起源”意味着所述生物样品在相同的动物模型(例如两只小鼠)上采样,并且所述样品是一致的(例如来自于同一肿瘤细胞谱系组织的两个活检组织,尿液的两个液体样品等)。
图2示出了使用msi为药物获得的相同起源(肿瘤组织)的两个组织切片的分子图谱。所述第一组织切片(图2a)在未与所述药物接触的动物中采样(对照,ctrl)。所述第二组织切片(图2b)在以前给药过所述药物的动物中采样(治疗过的,trt)。所述第一组织切片整体被用作第一roi。在所述第二组织切片中选择所述第二roi,并且所述第二roi对应于所述第二组织切片的其中所述分子被高度检出的区域。
在另一个实施方式中,从以前已暴露到剂量浓度与用于所述用药生物样品的剂量浓度不同的所述感兴趣分子的第二生物样品选择所述第二roi,所述第二生物样品来自于与所述用药生物样品相同的生物学起源。
图3示出了使用msi为药物获得的相同起源(肿瘤组织)的4个组织切片的分子图谱。所述第一组织切片在未与所述药物接触的动物中采样(对照,ctrl)。所有其他组织切片在以前给药过相同浓度的所述药物的动物中采样(trt1、trt2、trt3),但其中所述样品中的浓度是不同的。每个生物样品的roi是整个相应的组织切片。当然,可以将所述roi限制到每个组织切片的特定区域,例如表现出所述药物的最重要的浓度的roi。
在另一个实施方式中,从以前已暴露到同一个或第二感兴趣分子的第二生物样品选择所述第二roi,所述第二生物样品来自于与所述用药生物样品相同的起源。
图4示出了使用msi为两种不同药物获得的相同起源(肿瘤组织)的两个组织切片的分子图谱(图4a:药物a;图4b:药物b)。所述组织切片在以前给药过药物a和药物b中的任一者的动物中采样。在每个生物样品上手动分割所述roi1和roi2。为了了解药物a和b对所述生物标志物可能具有的药物效应,然后计算它在每个roi(1和2)中的强度/浓度,并为每个相应的roi绘图(图4c和4d)。
在特定实施方式中,本发明的方法可用于建立一种或多种感兴趣的分子与分子标志物之间的剂量效应曲线,其中roi(单一像素或几个像素)中感兴趣分子的每个强度或量根据同一roi中所述分子标志物的强度或量来表示。
图5示出了代表所述感兴趣化合物的浓度范围的生物样品的分子图谱。更高水平的图像分割允许获得多区段图谱(像素图)。每个像素含有以μg/g组织为单位的从c1至c11浓度的药物和生物标志物相关信息(图5a)。由此获得每个区段(像素)的药物生物效应,其对应于在所述药物的不同浓度或强度水平的基础上所述生物标志物的不同浓度或强度水平(图5b)。然后可以获得不同曲线,其给出在单个组织中的药物剂量效应。ed50:有效剂量中值,即实现50%的所需反应所需的剂量。
生物标志物分析
对每个所选roi进行分子分析,以便测量所述相应的生物样品的rio中一种或多种生物标志物的量或强度。
所述分子分析导致在分子水平上理解所述roi的组成,和/或检测并任选地定量一种或多种生物标志物。在有利情况下,所有roi均使用同一方法来分析。
根据一个实施方式,roi中所述生物标志物的量或强度的测量使用分子成像方法来进行。优选地,所述分子成像方法选自msi、ish、if、ihc、mri、成像质谱细胞仪、ct和pet成像。
在特定实施方式中,两个roi均通过msi进行分析。msi是指icp-ms、la-icpms、laesi、maldi、desi、sims、lesa和类似的表面提取策略、sims等。基本上,质谱成像(msi)能够直接从组织或流体样品同时记录数百种生物分子的分布,而不需标记且不需前期知识。可以使用基于基质辅助激光解吸/电离(maldi)的使用不同组织/流体制备程序的msi来分析蛋白质、肽、聚糖、脂质、代谢物和药物。
根据另一个实施方式,roi中所述生物标志物的量或强度的测量通过生物分析法来进行。优选地,所述生物分析法选自质谱、电泳、配体结合测定法、核磁共振、微透析和层析。
在特定实施方式中,在分析所述生物标志物之前从所述生物样品提取所述roi。在本发明的情形中,“roi的提取”意味着将所述roi与所述样品的其余部分在物理上分离。物理提取可以通过手动或利用激光的显微切割来进行。图6c示出了对组织切片进行的激光捕获显微切割(lcm)。由此对感兴趣的roi取样,并将其与所述组织切片的其余部分分离开。这种提取允许只在所述组织切片的必须考虑的部分上进行所述生物分析法,并以更高的准确度区分同一组织切片的不同部分。
图6示出了已暴露到药物的组织样品的分子图谱(图6a),然后对所述分子图谱进行手动或自动分割并鉴定所述roi(图6b),然后通过激光捕获显微切割进行roi提取(图6c)。
根据特定实施方式,在进行所述生物分析法之前将提取的roi在培养基中培养。
在特定实施方式中,在两个roi上进行生物分析,并通过在所述第一roi与第二roi(或其他的roi)之间比较基因和转录本表达、脂质组学、多肽组学、蛋白质组学和/或代谢组学的变化来评估所述感兴趣分子的效应。
所述roi的分子分析和结果的比较性研究可能产生不同的理解。
根据本发明,实施所述分子分析的方法可以是直接法或间接法(例如使用标签分子如抗体或rna序列)。例如,所述分子分析可以利用所述感兴趣区域的ihc、ish或fish成像、使用标记抗体的la-icp成像或用于检测组织上的金属的单独的la-icp成像、基因组分析、snips、lc-ms分析来进行。
在特定实施方式中,与所述感兴趣分子相关的生物标志物是已知的,并且所述分析步骤聚焦于所述生物标志物的检测和/或定量。本发明的方法可用于在非常精细的水平上鉴定所述靶组织。本发明的方法也可用于确定药物在靶组织中的预期效果的最佳剂量和/或用于筛选药物。
图1示出了其中与药物相关的生物标志物(bm)已知的实施方式。所述肿瘤组织的分子图谱显示出所述药物位于组织的特定部分上(图1b)。相应的roi中所述生物标志物的浓度的分析显示,与几乎不存在所述药物的roi相比,在所述药物集中的roi中所述bm的浓度降低(图1c)。本发明的方法允许区分同一组织切片的不同部分,而使用标准方法(lcms)的全局分析只显示出所述药物在整个组织切片中的全局影响。
图4示出了其中将本发明的方法用于筛选药物的实施方式。根据roi1和roi2中的药物水平,分析了与两种不同候选物(药物a相比于药物b)相关的预期效应(即生物标志物浓度的降低)。
图5示出了其中与药物相关的生物标志物(bm)已知的实施方式。执行本发明的方法以便从一个含有分析平行样并避免了生物学偏差的样品确定所述药物的剂量效应。不同像素中所述生物标志物的浓度的分析显示出所述组织中所述药物的浓度对所述生物标志物浓度的影响(图5b)。本发明的方法允许确定获得所需效果所需的药物的剂量或甚至是有效剂量中值(实现50%的所需反应所需的剂量)。正如在图5b中所示,对于50%的功效来说,药物b比药物a更加有效。相反,对于25%的功效来说,药物a比药物b更加有效。
根据另一个实施方式,进行所述roi的分析以鉴定与所述感兴趣分子相关的生物标志物。
质谱将关于所述感兴趣分子的生物学影响的分子信息提供为生命系统中脂质、代谢物、肽或蛋白质浓度的提高或降低。其他技术(if、ihc、elisa、ish、分光光度法或uv荧光)允许评估和定量使用标记抗体或与靶结合的核酸序列所靶向的肽、蛋白质、rna(核糖核酸)和dna(脱氧核糖核酸)。全部或一部分的这些实施方式可以组合,以显示出不同的感兴趣分子对同一生物样品的影响和/或不同浓度的感兴趣分子的影响和/或分子对不同的感兴趣生物样品的影响等。
根据本发明,可以使用多路成像技术(例如质谱成像),利用内源化合物将所述分子信息归一化,以分割与所述感兴趣分子相关的图像。这可能有助于将可变性降至最低。在这种情况下,使用外部或内部标准品化合物作为参比化合物。
应用
下面的实施例简要披露了本发明的方法的不同应用。在将药物给药到至少一个生物样品(例如体外试验、体内动物)后,使用成像技术来检测和定位所述药物。为药物获得所述样品的分子图谱。在强度的基础上分割所述分子图谱并选择两个感兴趣区域(roi)。
1-使用不同的分析方法来鉴定分子变化(例如使用质谱或质谱成像方法来靶向代谢物、dna、rna、蛋白质、脂质、肽、金属,或使用其他成像技术ish、ihc、mri、pet成像来比较所述两个感兴趣区域)。然后,鉴定在所述药物所在之处增加或减少的分子。可以对与所述药物相关的结果进行评分,并证实所述药物对某些细胞或器官子结构的影响。
2-手动或自动(使用激光显微切割仪器)提取所述roi,并使用分析方法来鉴定分子变化。使用生物分析方法例如质谱或其他技术如pcr来比较所述两个感兴趣区域的内含物。可以对与所述药物相关的结果进行评分,并证实所述药物对某些细胞或器官子结构的影响。
3-使用不同的分析方法鉴定分子变化,例如使用质谱或质谱成像来靶向代谢物、dna、rna、蛋白质、脂质、肽、金属,或使用其他成像技术ish、ihc、mri、pet成像来比较所述两个感兴趣区域。鉴定在所述药物所在之处增加或减少的分子。可以对非靶向化合物的结果进行评分,并证实所述药物对某些细胞或器官子结构的影响。
4-使用两个生物样品中的两种不同药物。选择两个roi,其对应于相应化合物在相应生物样品中的定位。使用不同的分析方法来鉴定分子变化,例如使用蛋白质组学、金属组学、转录组学、基因组学和代谢组学(包括脂质组学)的方法来比较所述两个感兴趣区域。然后,鉴定在所述药物所在之处增加或减少的分子。可以对靶向或非靶向化合物的结果进行评分,并评估在所述药物所在的特定区域中的剂量响应(功效或毒性)。
5-使用同一生物样品中的两种不同药物。使用相同的roi选择和使用分析工具的比较的过程,选择两个roi。使用不同的分析方法研究分子表达的差异。也可以选择两种药物所在的roi并研究它们对分子变化的影响和可能的协同效应。
6-可以作为图像像素产生更多的roi,以便从含有分析平行样并避免了生物学偏差的一个样品在药物浓度范围的基础上确定药物的剂量效应。所述不同像素内所述生物标志物的浓度的分析显示出所述组织中所述药物的浓度对所述生物标志物的浓度的影响。本发明的方法允许确定获得所需效果所需的药物的剂量或甚至是有效剂量中值(实现50%的所需反应所需的剂量)。
7-将生物样品在体外与将会渗透到细胞中并诱导分子相互作用和生理学变化的药物接触。使用相同的roi选择和使用分析工具的比较的过程,选择对应于不同细胞表型的潜在不同的roi,并分析对所述药物的相应反应(诱变、毒性等)。
实施例
吲哚胺-2,3-加双氧酶(ido1)是一种将色氨酸(trp)转变成犬尿氨酸(kyn)的酶。通过在微环境中降低trp并提高kyn水平而在肿瘤免疫逃逸中具有关键作用,ido1是免疫肿瘤学(i-o)领域中用于小分子药物发现的第一批靶点之一。已显示,强效且选择性的ido1抑制剂例如epacadostat(epa)通过恢复免疫系统健康而增强抗肿瘤活性。
在本研究中使用了定量质谱成像(qmsi),其目的不仅是定量代谢物和药物,而且还包括通过它们的组织学定位和定量来达到更高的信息水平。在这里报告了定量离体研究,其允许突出在特定肿瘤区域中epacadostat(epa)靶的暴露和药理效果功效。由于在作用位点处的暴露和向特定靶的暴露被鉴定为药物开发成功的最重要因素,因此本发明的目的是探索ido1抑制剂的靶暴露及其对肿瘤代谢的肿瘤内药效学效应。
1.材料和方法
1.1化学品和试剂
包括1,5-二氨基萘(1,5-dan)、2,5-二羟基苯甲酸(2,5-dhb)、犬尿氨酸-d4、色氨酸e-d5、甲酸和三氟乙酸在内的所有化学品购自sigma-aldrich(st.louis,mo)。犬尿氨酸-13c购自alsachim。甲醇、乙腈和optimalc-ms水购自fisherscientific(waltham,ma)。铟锡氧化物(ito)涂层的玻璃载片购自brukerdaltonics(bremen,germany)。
兔抗小鼠iggido第一抗体[#106134]和山羊抗兔iggh&l(hrp)第二抗体[#205718]以及通过辣根过氧化物酶然后通过steadydab/plus(棕色发色团)的检测系统均购自abcam(cambridge,uk)。
1.2样品收集和组织制备
将结肠癌ct26细胞系皮下移植到balb/c小鼠(charlesriverlaboratories,france)中。将小鼠随机分组,并在肿瘤具有70至120mm3的平均尺寸时开始治疗。将小鼠通过口饲管用100mg/kg的ido1抑制剂epacadostat(syngene,india)治疗,然后在2小时后处死。对肿瘤采样并在液氮中速冻15s。将样品保持在-80℃下直至使用。使用恒冷箱切片机(cm-3050s,leica,germany)并将切片机仓和样品夹冷冻在-23℃下,获得10微米厚的组织切片。将组织切片融化,安装在ito涂层的载片上用于下游maldi成像,并将superfrost载片上的连续切片用于组织学和免疫组织化学(ihc)分析。对一式两份生物平行样(远距离连续切片)进行分析,以保证分析的可重复性。对于lc-ms/ms分析来说,收获5个10μm厚的组织切片,用于制作校准曲线并进行epa、kyn和trp的定量。所有动物实验均符合2010/63/ue欧洲实验动物福利指令,并得到伦理委员会批准。
1.3epacadostat药物的定性和定量maldi分析
epa校准曲线分两步测量(大范围,然后是受限的范围)。然后,为使用至少10个点的浓度在10μm厚的对照肿瘤组织切片上制作的校准曲线确定检测下限(lod)、定量下限(lloq)和线性参数限度。epa的浓度范围是125至1pmol,其经验证为线性回归模型。在同一个ito载片上沉积校准曲线和感兴趣的组织切片(1个对照和3个治疗过的,一式两份)并进行分析。然后,使用htx-tm喷涂装置(htxtechnologies,llc,carrboro,nc),在肿瘤组织切片上沉积使用50/50acn/h2o基质制备的10mg/ml的过滤过的1,5-二氨基萘(1,5-dan)的均匀层。maldimsi分析使用带有smartbeamii激光器的7tmaldi-fticr(solarixxr,brukerdaltonics,bremen,germany)来进行。epacadostat的msi数据在casi负离子模式(m/z范围为436.0+/-30)下,使用在线校准以120μm的空间分辨率记录。数据采集使用来自于brukerdaltonics的flex软件(ftmscontrol2.1.0)进行。然后,使用multimaging软件(imabiotechsas)得到计算出的方程,并提取以pmol/mm2为单位的不同数量。然后在产生的感兴趣区域(roi)中获得数量向μg/g组织的转换。
1.4trp和kyn的定性和定量maldi分析
由于使用经典方法在ct26肿瘤模型中kyn不可检测并且trp略微可见,因此衍生策略是必需的。所述衍生反应相当于向中性分析物添加c11h16n+(作为x)单元。所述试剂使用自动喷雾器(suncollect,sunchrom)施加,并在室温下放置30min进行温育。然后使用htx-tm喷雾装置(htxtechnologies,llc,carrboro,nc),在所述肿瘤组织切片和校准曲线上喷涂使用50/50meoh/h2o+1%tfa基质制备的40mg/ml2,5-二羟基苯甲酸(2,5-dhb)与0.5μmkyn-d4衍生的内部标准品的混合物的均匀层。使用同位素修饰的化合物,如果在所述组织中存在,组织抑制效应应该允许获得相同的抑制效应。注意,使用氘代标准品(trp-d5和kyn-13c)执行所述校准曲线,以避免内源化合物的内部干扰。然后,使用带有smartbeamii激光器的7tmaldi-fticr(solarixxr,brukerdaltonics,bremen,germany)进行maldimsi分析。犬尿氨酸和色氨酸的msi数据在正离子模式(casi,m/z范围为350.0+/-150)下,使用在线校准以120μm的空间分辨率记录。数据采集使用来自于brukerdaltonics的flex软件(ftmscontrol2.1.0)进行。
1.5其他代谢物的maldi-fticr成像
对于其他代谢物的maldimsi来说,使用htx-tm喷涂装置(htxtechnologies,llc,carrboro,nc)在所述肿瘤组织切片上沉积用50/50acn/h2o基质制备的10mg/ml的过滤过的1,5-二氨基萘(1,5-dan)的均匀层。然后,使用带有smartbeamii激光器的7tmaldi-fticr(solarixxr,brukerdaltonics,bremen,germany)进行maldimsi分析。代谢物的msi数据在全扫描负离子模式(m/z范围100–1000)下,使用在线校准以120μm的空间分辨率记录。数据采集使用来自于brukerdaltonics的flex软件(ftmscontrol2.1.0)进行。
1.6lc-ms/ms分析
校准曲线(0至500nm)在含有5nm内部标准品(kyn-d4和trp-d5)的水中制备。然后,在含有10nm内部标准品的甲醇/水提取溶液中收集0.5至2mg之间的连续肿瘤切片。在4℃下进行过夜搅拌提取,然后在4℃下以3000×g离心15min,允许从kyn和trp两种校准曲线和所有切片回收上清液,用于lc-ms/ms分析。对于肿瘤来说,在分析之前在水中以1/2进行稀释。将总共5μl进样到lc-ms/ms系统中。不需要额外的过滤步骤。所述lc-ms/ms系统由超高效液相色谱–聚焦+lc系统构成,所述lc系统由rs柱温箱、rs泵和rs自动进样器(dionexultimate3000,thermofisherscientific)组成,含有偶联到tsqquantivathermoscientific(thermofisherscientific)的waters柱(cortecsc1875×3mm,粒径=2.7μ)。数据采集和处理使用tsqquantiva软件2.01292版和xcalibur3.0软件来进行。
1.7msi数据处理和分析
1.2软件(imabiotechsas)。这个多模态成像平台将定量质谱成像(qmsi)和显微镜平台与统计分析相组合,用于在细胞水平上理解组学信息。msi数据从每个组织切片以及与所述组织切片相邻的基质对照区域获取,以检查样品制备期间分析物的分散/移位。因此,与epa和/或kyn存在相关的roi通过图像分割算法给出。首先,所述算法在分子信号阈值的基础上将所述样品分成不同类别(在本研究的情况下,对于epa和kyn两者来说为2个类别或roi)。然后,所述算法对所述两个roi进行平滑处理,以将它们转换为连通空间。最后,使用multimaging软件,使用下述公式计算暴露评分:
其中nroi是roi内的像素数目,n总是整个样品内的像素数目。
1.8免疫组织化学分析、数字扫描图像和高清晰度叠加
使用购自abcam(cambridge,uk)并适合于新鲜冷冻组织切片的兔多克隆抗体对连续切片进行染色,用于ido1组织学定位。首先将切片暴露到0.5%triton-x,在室温下15min,并用磷酸盐缓冲盐水清洗,然后添加抗ido1第一抗体(1:50稀释),并用山羊抗兔iggh&l(hrp)第二抗体(1:2000)处理。检测系统是通过辣根过氧化物酶,然后是稳态dab/plus(棕色发色团)。
在msi数据采集后,使用100%甲醇清洗除去任何残留的基质,然后将所述组织样品用苏木精和曙红(h&e)溶液染色。然后使用数字载片扫描仪(3dhistechpannoramic)记录h&e染色或ihc的高分辨率组织学图像,然后载入multimaging技术,以使用复杂的分子图像进行高清晰度叠加。
2.结果
2.1epacadostat药物检测、定量和组织学定位
进行了qmsi和lc-ms/ms分析以定量ct26组织、血浆和血液样品中epacadostat的绝对水平。首先,为epaqmsi分析进行了样品制备和开发方法。在对照ct26组织切片上点样校准曲线,允许获得显示出12至870μg/g的线性范围的校准曲线(r2=0.996),检测限(lod)为12μg/g,定量下限(lloq)为15μg/g(图7a)。然后,在一个对照和三个治疗过的切片各一式两份中显示了分子图像,其显示出第一epa同位素(m/z435.9844)的组织学定位。然后使用得到的校准曲线进行qmsi,对于三个生物学平行样给出38至53μg/g之间的量(图8a)。也对ct26连续组织切片进行lc-ms/ms定量。图7b示出了以nm为单位获得的epa校准曲线。当使用lc-ms/ms相比于qmsi时,平均epa量分别为38.5μg/g相比于46μg/g(图8b),显示出两种技术之间17%的变差。还对血浆和血液进行了epa定量(图8c)。
2.2靶暴露分析
对于靶暴露分析来说,首先,epa信号的自动分割突出了两个roi(1和2)。然后对epa绝对定量进行评估(使用qmsi),并从三个区域自动提取(高epa的roi1,低epa的roi2和整个治疗过的肿瘤的roi3)。结果显示在roi1中的量为68μg/g,在roi2中的量为25μg/g,并且在整个切片(roi3)中的量为42μg/g;这意味着几乎61%的epa信号(26μg/g)集中在整个组织切片的38%(roi1)内,并且39%(15μg/g)的信号位于62%的剩余肿瘤区域(roi2)中(图9a)。然后,通过免疫染色突出了ido1酶表达的强度。因此,所述半定量分析允许区分显示出不同ido1表达水平的两个区域,从最高表达(1)到最低表达(2)(图9b)。有趣的是,也注意到ido1表达水平(通过组织学进行的半定量评估)与epa水平之间的正相关性。
2.3kyn和trp代谢物的药效学研究
对于内源代谢物来说,优化的衍生步骤允许检测和定量trp和kyn两者,同时提高它们的检测灵敏度。使用msi分析显示了epacadostat、trp和kyn在ct26肿瘤上的组织学定位(图10a)。分子图像显示,与治疗过的组织相比,对照ct26组织上kyn水平更高并且trp水平更低。随后,对于qmsi来说,使用相同的衍生策略进行归一化的trp和kyn两者的校准曲线(图11a)。在对照和治疗过的ct26组织切片上使用qmsi和lc-ms/ms分析对kyn和trp两者进行绝对定量,然后将kyn/trp比率计算作图在图9b上,其显示出两种技术之间良好的相关性。注意到在epa治疗后kyn/trp比率降低,并且不论使用何种技术均发现6倍的降低。还分析了血浆和血液样品,结果显示在治疗时kyn/trp比率降低3倍(图11b)。
2.4从靶暴露到反应功效分析:epa在ct26肿瘤小鼠模型上的区域性/药理效果
通过作为ido1酶的直接酶产物的kyn代谢物的绝对定量来跟踪epa药物的药理功效。三个roi中的绝对kyn量显示15%的kyn存在于38%的整个肿瘤(roi1)中,85%的kyn存在于roi2中(图12a)。最后,乳酸代谢物与epa和kyn的组织学分布示出在图12b中,然后将从高和低epa区域(分别为1和2)的每个x和y位置提取的所有分子的相对强度作图。由此发现了epa、kyn和乳酸代谢物之间的负相关性,因为roi1上的高epa水平对应于kyn和乳酸两者的低表达水平(图12c)。
结果分析
研究结果显示,在血浆和血液中的量分别在6.6+/-1μg/g和7.3+/-2μg/g之间(图7c)。然后,对治疗过的ct26肿瘤使用lc-ms/ms和qmsi,组织上epa的可用量比血浆高3或4倍(在35至48μg/g之间),两种技术之间的变差低于20%。与epa抑制效应相关,在血浆中注意到犬尿氨酸水平以剂量依赖性方式降低。在这个实验中,观察到约24小时>50%的抑制,在这一整天的过程中剂量为100mg/kg。
我们最后的研究显示出在p815小鼠模型上kyn水平从25-34μg/g(p815高ido1)降低到<60ng/g(p815低ido1)。
在本研究中,公知ct26鼠类结肠癌细胞表达ido1,因此将其用于确定ido1抑制对肿瘤生长的影响。事实上,ct26模型已被良好地用于ido1抑制剂的药理评估。在历史上,药物的丰度和分布通过已确立的技术例如定量全身放射自显影(qwba)或组织匀浆和lc-ms/ms分析来评估。然而,qwba不能区分活性药物与其代谢物,而lc-ms/ms尽管高度灵敏,但不能报告空间分布。质谱成像(msi)可以区分药物及其代谢物和内源化合物,并同时报告它们的分布。msi数据正影响药物开发,并且目前被用于诸如dmpk、pd和毒性领域中的调查性研究。
本研究显示,对于靶暴露研究来说,使用qmsi技术具有重大影响。在比较对照和治疗过的ct26肿瘤时,分割出与内部包含的epa量相关的特定区域。由此确认肿瘤向epa的暴露,然后提取出两个可区分的区域(1为高epa,2为低epa)。对应于68μg/g的差不多61%的epa药物位于38%的整个肿瘤中。ido1酶的半定量分析显示出与roi2相比,在roi1中ido1高表达,这支持了epa向其ido1靶的暴露。
由于kyn和trp的基础水平相当低,因此启动了衍生步骤以允许内源代谢物的组织上定位。由于maldimsi被用于广泛质量范围内的多样化分析物的多重检测,因此分析物覆盖率高度受限于更丰富的化合物。组织上或组织中分析物化学衍生通过选择性标记特异性针对分析物类别的官能团,并在同时改变它们的分子质量并提高它们的电离效率以及因此它们的灵敏度,解决了这些问题。因此,trp和kyn的灵敏度提高,允许对它们进行组织上分布和定量研究。几项研究显示,与来自于正常志愿者的血清相比,来自于癌症患者的血清具有更高的kyn/trp比率,与ido1活性的提高相符。使用qmsi和lc-ms/ms两者,我们的数据显示,当将对照与治疗过的ct26组织的血浆和血液相比时,kyn/trp比率分别降低6倍和3倍(图11b)。
同样,最近验证了kyn/trp比率作为许多癌症中的预后工具:宫颈癌,成胶质细胞瘤和肺癌,其中将lc-ms/ms用于代谢物定量。然后,与不存在epa的区域(对照肿瘤)相比,提取kyn表达,显示出epa存在与其对kyn的药理效果之间的区域相关性。
最后,实现了ido1酶免疫染色,允许观察epa靶的组织学定位。在与ido1抑制相关的底物和产物变化之外,癌细胞还表现出代谢变化,这将它们与健康组织区分开,并使它们的代谢过程对药理靶向敏感。细胞代谢物具有多种多样的物理化学性质,因此它们的检测和定量需要各种不同分析方法的组合。特定代谢物的存在可能指示肿瘤的代谢状态;然而,并不总是能够仅从代谢物丰度的测量直接推断出特定代谢途径的活性。
本研究的结果显示出乳酸与epa分子之间的组织学反定位。由于乳酸是糖酵解途径的最终产物,因此在epa高度集中的同一组织学区域中它的减少对应于显著的糖酵解降低。代谢物例如乳酸和葡萄糖的区域性水平的评估,使用用于脑缺血快速冷冻活检样品的定量生物发光成像来进行。
在各种不同的肿瘤和癌症类型中,乳酸被证明是一种预后指示物,因为在人类宫颈癌、头颈癌、高等级胶质细胞瘤和非小细胞肺癌中,它的水平升高与更加不良的患者预后、不良的无病或无转移生存期和不良的总生存期相关。这个特点使得乳酸代谢成为进一步研究的兴趣所在,其不仅作为生物标志物,而且作为潜在的治疗终点或靶点。
随后,通过在肿瘤细胞中抑制ido1和减少kyn,epa恢复了各种不同细胞的增殖、活化和调控。事实上,这将支持epa的药理效果。此外,还使用maldimsi突出了tca循环和能量途径代谢,显示出不同的组织学定位和相对强度(数据未示出)。一组特定代谢物的存在可以指示肿瘤的代谢状态。实现了细胞外肿瘤微环境表征,显示出糖酵解的特征例如乳酸、葡萄糖、苹果酸、柠檬酸、谷氨酰胺和脯氨酸。普遍的情况是,在肿瘤中,更高比例的葡萄糖碳从线粒体转移出去并转变成乳酸。此外,还显示了谷氨酰胺代谢和与脯氨酸合成相关的功能。最后,还显示了嘌呤核苷酸的降解。
3.结论
总而言之,这些结果报告了一项定量超高质量分辨率研究,其允许突出epa的原位pk/pd研究。事实上,实现了不同的研究:
■通过组织上药物定量来进行生物分布和生物利用度研究。
■在生物标志物和/或药物组织学定位的基础上进行组织分割后进行靶向组织暴露研究。
■还进行了药效学研究,其显示出药物向反应效应的区域性暴露,这使功效分析成为可能。
事实上,由于在作用位点处和向其特定靶点的暴露被鉴定为是药物发现和化学探针设计成功的最重要因素,因此,这些结果显示并证实了qmsi以及更广义来说msi对研究靶占有率与药物功效之间的关系的巨大贡献。
- 还没有人留言评论。精彩留言会获得点赞!