地榆药材的UPLC指纹图谱构建方法和检测方法与流程
本发明涉及中药质量控制
技术领域:
,特别涉及地榆药材的uplc指纹图谱构建方法和检测方法。
背景技术:
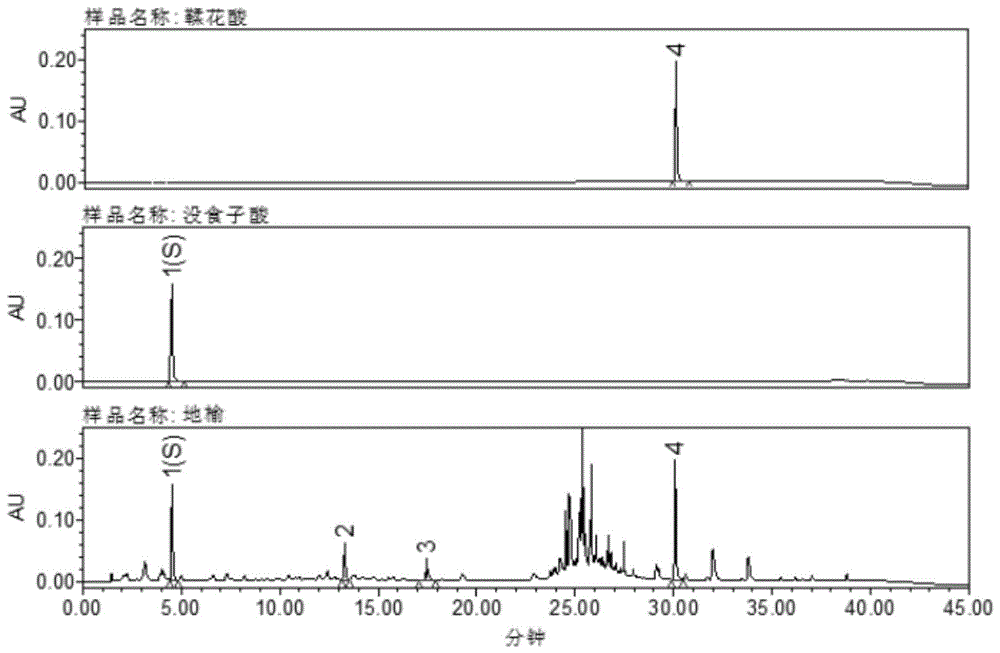
:《中国药典》2020年版记载地榆为蔷薇科植物地榆sanguisorbaofficinalisl.或长叶地榆sanguisorbaofficinalisl.var.longifolia(bert.)yüetli的干燥根。后者习称“绵地榆”。地榆味苦、酸、涩,微寒;归肝、大肠经;具有凉血止血,解毒敛疮,可用于治疗便血,痔血,血痢,崩漏,水火烫伤,痈肿疮毒等。通常,地榆需要春季将发芽时或秋季植株枯萎后采挖,采挖后需依次经过除去须根,洗净,干燥,或趁鲜切片,干燥等加工工序,地榆不同的生长地、不同的处理工艺等均会导致地榆药材质量的差异,故而很难评价相应的地榆药材是否满足药用标准。因此,亟需构建一个准确且稳定的地榆药材质量评价标准。技术实现要素:基于此,有必要提供一种地榆药材的uplc指纹图谱构建方法和检测方法。该方法构建得到的地榆药材指纹图谱可以用于对地榆药材进行质量评价,提高地榆药材检测的准确性和稳定性。一种构建地榆药材指纹图谱的方法,包括以下步骤:配制没食子酸对照品溶液、地榆对照药材溶液和多批地榆供试品溶液;采用超高液相色谱法检测所述没食子酸对照品溶液、所述地榆对照药材溶液和各批地榆供试品溶液,获得对应的色谱图;根据地榆对照药材的色谱图建立地榆药材对照特征图谱;标识各地榆供试品的色谱图中的共有峰,从所述共有峰中选择与所述地榆对照药材保留时间一致的峰,作为特征峰,并使所述特征峰与所述地榆药材对照特征图谱相应的各特征峰对应,构建所述地榆药材指纹图谱。在其中一些实施例中,采用以下方法建立所述地榆药材对照特征图谱:采用中药色谱指纹图谱相似度评价系统,以没食子酸对应的峰为参照峰,计算地榆对照药材的各特征峰的相对保留时间及相对峰面积,按照平均数法或中位数法生成对照图谱,建立地榆药材对照特征图谱。在其中一些实施例中,所述超高效液相色谱仪的流动相a相为甲醇,b相为0.08%-0.12%的磷酸的水溶液。在其中一些实施例中,所述超高效液相色谱仪的洗脱程序为:0-6min,a相从3%到6%,b相从97%到94%;6-8min,a相从6%到11%,b相从94%到89%;8-20min,a相为11%,b相为89%;20-25min,a相从11%到42%,b相从89%到58%;25-30min,a相为42%,b相为58%;30-35min,a相为42%到55%,b相为58%到45%;35-40min,a相从55%到97%,b相从45%到3%;40-45min,a相为97%,b相为3%;45-46min,a相从97%到3%,b相从3%到97%;46-60min,a相为3%,b相为97%。在其中一些实施例中,流速为0.1ml/min-0.3ml/min,检测波长为270nm。在其中一些实施例中,所述特征峰包含第一峰、第二峰、第三峰和第四峰中至少三个峰;所述第一峰的保留时间为4.49-4.67min,所述第二峰的保留时间为13.28-13.50min,所述第三峰的保留时间为17.53-17.70min,所述第四峰的保留时间为30.37-30.71min。在其中一些实施例中,所述特征峰包含第一峰、第二峰、第三峰和第四峰中至少三个峰;其中,所述第一峰为具有以下质谱信息的峰:正离子模式下的m/z为171.0288±3ppm、或负离子模式的m/z为169.0132±3ppm;所述第二峰为具有以下质谱信息的峰:正离子模式下的m/z为315.0710±3ppm、或负离子模式下的m/z为345.0829±3ppm;所述第三峰为具有以下质谱信息的峰:正离子模式下的m/z为291.0861±3ppm、或负离子模式的m/z为289.0720±3ppm;所述第四峰为具有以下质谱信息的峰:负离子模式的m/z为300.9991±3ppm。在其中一些实施例中,所述第一峰的m/z为171.0288±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:153.0183±3ppm、127.0391±3ppm、125.0235±3ppm、109.0288±3ppm、107.0132±3ppm、81.0341±3ppm;或所述第一峰的m/z为169.0132±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:125.0232±3ppm;或所述第二峰的m/z为315.0710±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:153.0182±3ppm;或所述第二峰的m/z为345.0829±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:239.0594±3ppm、148.0155±3ppm、223.0280±3ppm、87.0438±3ppm;或所述第三峰的m/z为291.0861±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:139.0390±3ppm、123.0442±3ppm、147.0441±3ppm;或所述第三峰的m/z为289.0720±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:109.0282±3ppm、125.0232±3ppm、123.0440±3ppm、151.0390±3ppm;或所述第四峰的m/z为300.9991±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:229.0138±3ppm。在其中一些实施例中,所述第一峰为没食子酸对应的峰,所述第三峰为儿茶素或表儿茶素对应的峰,所述第四峰为鞣花酸对应的峰。在其中一些实施例中,所述没食子酸对照品溶液的浓度为0.8mg/ml-1.2mg/ml。在其中一些实施例中,所述地榆供试品溶液的浓度为3mg/ml-6mg/ml。一种地榆药材的检测方法,包括以下步骤:配制地榆药材的供试品溶液;采用超高效液相色谱-质谱联用技术对所述供试品溶液进行检测,获得相应的谱图;采用上述方法构建而成的地榆药材指纹图谱对所述谱图进行分析。在其中一些实施例中,所述超高效液相色谱仪的色谱条件为:a相为甲醇,b相为0.08%-0.12%的甲酸的水溶液;洗脱程序为:0-6min,a相从3%到6%,b相从97%到94%;6-8min,a相从6%到11%,b相从94%到89%;8-20min,a相为11%,b相为89%;20-25min,a相从11%到42%,b相从89%到58%;25-30min,a相为42%,b相为58%;30-35min,a相为42%到55%,b相为58%到45%;35-40min,a相从55%到97%,b相从45%到3%;40-45min,a相为97%,b相为3%;45-46min,a相从97%到3%,b相从3%到97%;46-60min,a相为3%,b相为97%;质谱条件为:hesi离子源,鞘气流速30-40arb,辅助气流速为8-12arb,喷雾电压为3-4kv,s-lens电压为45-55v,加热温度为340℃-360℃,毛细管温度为340℃-360℃,正、负离子模式下分别扫描采集,扫描质量范围为m/z100-1200,归一化碰撞能量分别为20、40和60ev。。一种地榆药材的检测方法,包括以下步骤:(1)配制地榆药材的供试品溶液和对照品溶液;(2)采用超高效液相色谱-质谱联用技术分别检测所述供试品溶液和对照品溶液,获得地榆药材的供试品图谱和对照品图谱;以没食子酸、儿茶素(表儿茶素)和鞣花酸中至少两种组分对应的峰为特征峰,根据所述供试品图谱和对照品图谱,对地榆药材的供试品的质量进行分析。本发明具有以下有益效果:本发明的通过采用uplc法对各批次地榆供试品进行检测分析,寻找出各地榆供试品的特征峰,构建指纹图谱,为地榆药材的质量控制奠定了基础。且本发明简单、可靠、重现性好,能够较为全面地反应地榆药材的整体质量,进而能够有效地提高地榆药材质量检测的准确性和稳定性。附图说明图1为地榆特征图谱与对照品图谱对照图;图2为地榆对照药材的特征图谱;图3为基于图2所建立的地榆药材对照特征图谱;图4为23批地榆供试品药材的特征图谱;图5为地榆供试品溶液总离子流图及紫外吸收色谱图;图6为1号峰一级质谱扫描图(正离子模式);图7为1号峰一级质谱扫描图(负离子模式);图8为m/z171.0288峰二级质谱图(正离子模式);图9为m/z169.0132峰二级质谱图(负离子模式);图10二级离子碎片质谱镜像图(负离子模式);图11为没食子酸结构式;图12为m/z171.0288峰可能的裂解方式(正离子模式);图13为m/z169.0132峰可能的裂解方式(负离子模式);图14为2号峰一级质谱扫描图(正离子模式);图15为2号峰一级质谱扫描图(负离子模式);图16为m/z315.0710峰的二级质谱图(正离子模式);图17为m/z345.0829峰的二级质谱图(负离子模式);图18为3号峰一级质谱扫描图(正离子模式);图19为3号峰一级质谱扫描图(负离子模式);图20为m/z291.0861峰的二级质谱图(正离子模式);图21为m/z289.0720峰的二级质谱图(负离子模式);图22为二级离子碎片质谱镜像图(正离子模式),其中,上为m/z291.0861峰二级碎片;下为数据库中儿茶素;图23为二级离子碎片质谱镜像图(负离子模式),其中,上为m/z289.0720峰二级碎片;下为数据库中儿茶素);图24为儿茶素结构式(c15h14o6);图25为m/z290.0861峰可能的裂解方式(正离子模式);图26为m/z289.022峰可能的裂解方式(负离子模式);图27为4号峰峰一级质谱扫描图(负离子模式);图28为m/z300.9991峰的二级质谱图;图29为二级离子碎片质谱镜像图(负离子模式),其中,上为m/z300.9991峰二级碎片;下为数据库中鞣花酸二级碎片;图30为鞣花酸结构式(c14h6o8)。具体实施方式为了便于理解本发明,下面将对本发明进行更全面的描述,并给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的
技术领域:
的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。本发明一实施方式提供了一种构建地榆药材指纹图谱的方法,包括以下步骤:s101:配制没食子酸对照品溶液、地榆对照药材溶液和多批地榆供试品溶液。在其中一些实施例中,没食子酸对照品溶液的浓度为0.8mg/ml-1.2mg/ml;进一步地,没食子酸对照品溶液的浓度为0.90mg/ml、0.95mg/ml、1.00mg/ml、1.05mg/ml、1.10mg/ml、1.15mg/ml或1.2mg/ml。在一些实施例中,没食子酸对照品溶液的溶剂为甲醇。在一些实施例中,地榆供试品溶液的浓度为3mg/ml-6mg/ml;进一步地,地榆供试品溶液的浓度为3.5mg/ml、3.8mg/ml、3.9mg/ml、4.0mg/ml、4.1mg/ml、4.2mg/ml、4.5mg/ml、5.0mg/ml、5.5mg/ml或6mg/ml。在一些实施例中,地榆对照药材溶液的浓度为3mg/ml-6mg/ml;进一步地,地榆对照药材溶液的浓度为3.5mg/ml、3.8mg/ml、3.9mg/ml、4.0mg/ml、4.1mg/ml、4.2mg/ml、4.5mg/ml、5.0mg/ml、5.5mg/ml或6mg/ml。在一些实施例中,地榆对照药材溶液和地榆供试品溶液的配制方法为:(1)分别称取预定量的地榆对照药材或地榆供试品;(2)分别将地榆对照药材和地榆供试品置于溶剂中,加热回流,冷却,定容至所需体积,过滤,取滤液。在一些实施例中,地榆对照药材溶液和地榆供试品溶液的溶剂为10%-40%的甲醇;进一步地,溶剂为25%-35%的甲醇;进一步地,溶剂为30%的甲醇。在一些实施例中,步骤s101中,多批地榆供试品的批次至少为15批;进一步地,至少20批。在一些实施例中,各批地榆供试品为不同产地的地榆供试品。s102:采用超高液相色谱法检测没食子酸对照品溶液、地榆对照药材溶液和各批地榆供试品溶液,获得对应的色谱图。在一些实施例中,超高效液相色谱仪(uplc)的流动相a相为甲醇,b相为0.08%-0.12%的磷酸的水溶液。在一些实施例中,色谱柱为waterscortecst3(150mm×2.1mm,1.6μm)色谱柱。在一些实施例中,uplc的流速为0.1ml/min-0.3ml/min;进一步地,流速为0.25ml/min。在一些实施例中,洗脱程序为:0-6min,a相从3%到6%,b相从97%到94%;6-8min,a相从6%到11%,b相从94%到89%;8-20min,a相为11%,b相为89%;20-25min,a相从11%到42%,b相从89%到58%;25-30min,a相为42%,b相为58%;30-35min,a相为42%到55%,b相为58%到45%;35-40min,a相从55%到97%,b相从45%到3%;40-45min,a相为97%,b相为3%;45-46min,a相从97%到3%,b相从3%到97%;46-60min,a相为3%,b相为97%。s103:根据地榆对照药材的色谱图建立地榆药材对照特征图谱;标识各地榆供试品的色谱图中的共有峰,从共有峰中选择与地榆对照药材保留时间一致的峰,作为特征峰,并使特征峰与地榆药材对照特征图谱相应的特征峰对应,构建地榆药材指纹图谱。进一步地,步骤s103中,采用以下方法建立地榆药材对照特征图谱:采用中药色谱指纹图谱相似度评价系统,以没食子酸对应的峰为参照峰,计算地榆对照药材的各特征峰的相对保留时间及相对峰面积,生成对照图谱,建立地榆药材对照特征图谱。更进一步地,通过采用中药色谱指纹图谱相似度评价系统,按平均数法(或中位数法)生成对照图谱,如此可以建立地榆药材对照特征图谱。可理解的,本发明中的“中药色谱指纹图谱相似度评价系统”应理解为具有与该系统相同或相似功能的系统,只要能够实现本发明所述功能,不与本发明的发明目的相悖即可,应理解为均在本发明的保护范围内。在一些实施例中,特征峰包含第一峰、第二峰、第三峰和第四峰中至少三个。在一些实施例中,第一峰的保留时间为4.49-4.67min,第二峰的保留时间为13.28-13.50min,第三峰的保留时间为17.53-17.70min,第四峰的保留时间为30.37-30.71min。在一些实施例中,第一峰为具有以下质谱信息的峰:正离子模式下的m/z为171.0288±3ppm,负离子模式的m/z为169.0132±3ppm。在一些实施例中,第一峰的m/z为171.0288±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:153.0183±3ppm、127.0391±3ppm、109.0288±3ppm;进一步地,第一峰的m/z为171.0288±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:153.0183±3ppm、127.0391±3ppm、125.0235±3ppm、109.0288±3ppm、107.0132±3ppm、81.0341±3ppm;进一步地,第一峰的m/z为171.0288±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:153.0183±3ppm、127.0391±3ppm、125.0235±3ppm、109.0288±3ppm、107.0132±3ppm、81.0341±3ppm。进一步地,第一峰的m/z为169.0132±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:125.0232±3ppm。在一些实施例,第一峰为没食子酸对应的峰。在一些实施例,第二峰为具有以下质谱信息的峰:正离子模式下的m/z为315.0710±3ppm、或负离子模式下的m/z为345.0829±3ppm;进一步地,第二峰的m/z为315.0710±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:153.0182±3ppm。进一步地,第二峰的m/z为345.0829±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:239.0594±3ppm。进一步地,第二峰的m/z为345.0829±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:239.0594±3ppm、148.0155±3ppm。进一步地,第二峰的m/z为345.0829±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:239.0594±3ppm、148.0155±3ppm、223.0280±3ppm、87.0438±3ppm。在一些实施例,第三峰为具有以下质谱信息的峰:正离子模式下的m/z为291.0861±3ppm,负离子模式的m/z为289.0720±3ppm。进一步地,第三峰的m/z为291.0861±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:139.0390±3ppm、123.0442±3ppm。进一步地,第三峰的m/z为291.0861±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:139.0390±3ppm、123.0442±3ppm、147.0441±3ppm。进一步地,第三峰的m/z为289.0720±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:109.0282±3ppm或125.0232±3ppm。进一步地,第三峰的m/z为289.0720±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:109.0282±3ppm、125.0232±3ppm、151.0390±3ppm。进一步地,第三峰的m/z为289.0720±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:109.0282±3ppm、125.0232±3ppm、123.0440±3ppm、151.0390±3ppm。进一步地,第三峰为儿茶素或表儿茶素(同分异构体)对应的峰。在一些实施例中,第四峰为具以下质谱信息的峰:负离子模式的m/z为300.9991±3ppm。进一步地,第四峰的m/z为300.9991±3ppm的峰的二级质谱图至少在以下m/z存在碎片离子峰:229.0138±3ppm。进一步地,第四峰为鞣花酸对应的峰。本发明技术人员,创新性地选择上述四个峰中至少三个作为特征峰,构建地榆药材指纹图谱,如此其能够较为全面的反应地榆药材的整体情况,当该地榆药材指纹图谱用于进行地榆药材的质量检测时,更能全面地检测地榆药材的信息,进而提高质控的准确性。本发明一实施方式提供了一种地榆药材的检测方法,包括以下步骤:s210:配制榆药材的供试品溶液;步骤s210中供试品溶液的配制同步骤s101,在此不再进行赘述。s220:采用超高效液相色谱-质谱联用技术对供试品溶液进行检测,获得相应的谱图;在一些实施例中,采用uplc-qtof/ms方法。在一些实施例中,超高效液相色谱仪(uplc)的流动相a相为甲醇,b相为0.08%-0.12%的甲酸溶液。在一些实施例中,色谱柱为waterscortecst3(150mm×2.1mm,1.6μm)色谱柱。在一些实施例中,uplc的流速为0.1ml/min-0.3ml/min;进一步地,流速为0.25ml/min。在一些实施例中,洗脱程序为:0-6min,a相从3%到6%,b相从97%到94%;6-8min,a相从6%到11%,b相从94%到89%;8-20min,a相为11%,b相为89%;20-25min,a相从11%到42%,b相从89%到58%;25-30min,a相为42%,b相为58%;30-35min,a相为42%到55%,b相为58%到45%;35-40min,a相从55%到97%,b相从45%到3%;40-45min,a相为97%,b相为3%;45-46min,a相从97%到3%,b相从3%到97%;46-60min,a相为3%,b相为97%。在一些实施例中,质谱条件为:hesi离子源,鞘气流速30-40arb,辅助气流速为8-12arb,喷雾电压为3-4kv,s-lens电压为45-55v,加热温度为340℃-360℃,毛细管温度为340℃-360℃,正、负离子模式下分别扫描采集,扫描质量范围为m/z100-1200,归一化碰撞能量分别为20、40和60ev。。进一步地,质谱条件为:hesi离子源,鞘气流速35arb,辅助气流速为10arb,喷雾电压为3.80kv,s-lens电压为50v,加热温度为350℃,毛细管温度为360℃。进一步地,步骤s220中,包括采用超高效液相色谱-质谱联用技术获取的各特征峰的质谱的步骤,各特征峰的质谱信息如上述,在此不再进行赘述。s230:采用上述方法构建而成的地榆药材指纹图谱对谱图进行分析。步骤s230中的地榆药材指纹图谱的构建方法如上所述,在此不再进行赘述。可理解的,步骤s230中,可以根据需要(如定性和/或定量),确定不同的检测指标(如保留时间和/或峰面积等),在此不进行特别限定,应理解为均在本发明的保护范围内。可理解的,步骤s230中可以采用现有的分析方法来进行定量,如内标或外标法,应理解为均在本发明的保护范围内。另外,还可以包括检测各峰质谱信息的步骤。下面列举具体实施例来对本发明进行说明。以下实施例中仪器和试剂来源如下:仪器:thermo高效液相色谱仪(vanquish,themro公司);waters高效液相色谱仪(h-class,waters公司);waterscortecst3(150mm×2.1mm,1.6μm)色谱柱;万分之一分析天平(me204e,梅特勒-托利多公司);百万分之一分析天平(xp26,梅特勒-托利多公司);数控超声波清洗器(kq500d,昆山市超声仪器有限公司);恒温水浴锅(hws28型,上海一恒科技有限公司型号);超纯水系统(milli-qdirect,默克股份有限公司)。试剂:乙醇(天津市富宇精细化工有限公司,分析纯);甲醇(天津市富宇精细化工有限公司,分析纯);磷酸(天津市科密欧化学试剂有限公司,色谱纯);乙腈(默克股份有限公司,色谱纯);水为超纯水(实验室自制)。试药:没食子酸(中国食品药品检定研究院,含量:90.8%,批号:110831-201204);鞣花酸(中国食品药品检定研究院,含量:88.8%,批号:111959-201903);地榆对照药材(中国食品药品检定研究院,批号:121286-201703);23批地榆药材经广东一方制药有限公司魏梅主任药师鉴定为蔷薇科植物地榆sanguisorbaofficinalisl.的干燥根。产地及编号见表1。表1样品信息表序号产地序号产地s1甘肃省定西市陇西县s13山东省日照市s2甘肃省定西市陇西县s14山东省日照市s3甘肃省定西市陇西县s15山东省临沂市s4甘肃省定西市陇西县s16山东省临沂市s5甘肃省定西市陇西县s17山东省日照市s6甘肃省定西市陇西县s18甘肃省陇西县s7甘肃省定西市陇西县s19甘肃省陇西县s8甘肃省定西市陇西县s20甘肃省陇西县s9甘肃省定西市陇西县s21甘肃省平凉市s10湖南郴州桂阳s22甘肃省平凉市s11湖南郴州桂阳s23甘肃省平凉市s12湖南郴州实施例11.1色谱条件采用waterscortecst3(150mm×2.1mm,1.6μm)色谱柱;以甲醇为流动相a,以0.1%磷酸溶液为流动相b,按下表中的规定进行梯度洗脱;流速为每分钟0.25ml;柱温为35℃;检测波长为270nm。1.2对照品溶液的配制分别取没食子酸、鞣花酸对照品适量,精密称定,加甲醇制成每1ml含1mg的溶液。1.3供试品溶液的制备取地榆粉末(过四号筛)约0.1g,精密称定,置具塞锥形瓶中,精密加30%甲醇25ml,称定重量,加热回流60分钟,放冷,再称定重量,用30%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。1.4方法学考察1.4.1精密度试验取同一批地榆样品(s1),按照“1.3”项下方法制备供试品溶液,按“1.1”项色谱条件重复进样测定6次,以没食子酸为参照峰,计算4个共有峰的相对保留时间及相对峰面积的rsd。结果表明,4个色谱峰的相对保留时间及相对峰面积rsd均小于3%,说明一起精密度良好。1.4.2重现性试验取同一批地榆样品(s1),按照“1.3”项下方法平行制备6份供试品溶液,按“1.1”项色谱条件重复进样测定6次,以没食子酸为参照峰,计算4个共有峰的相对保留时间及相对峰面积的rsd。结果表明,4个色谱峰的相对保留时间及相对峰面积rsd均小于3%,表明本方法重现性良好。1.4.3稳定性试验取同一供试品溶液,按“1.1”项色谱条件,分别于0,2,4,6,8,12,24h进样,以没食子酸为参照峰,计算4个共有峰的相对保留时间及相对峰面积的rsd。结果表明,4个色谱峰的相对保留时间及相对峰面积rsd均小于3%,表明供试品在24h内稳定。1.4.4专属性考察取没食子酸对照品溶液、鞣花酸对照品溶液、地榆药材供试品溶液,按“1.1”项色谱条件注入液相色谱仪,记录色谱图。结果显示地榆中特征图谱没食子酸峰、鞣花酸峰与参照物溶液的保留时间一致(参见图1),本方法具有良好的专属性。1.5特征图谱的建立1.5.1指纹图谱共有模式的建立取地榆对照药材(批号:121286-201703)及23批地榆药材,按“1.1”项下色谱条件与“1.3”项下确定的供试品溶液制备方法,进样测定,得到地榆对照药材特征图谱如图2,使用《中药色谱指纹图谱相似度评价软件》,以没食子酸峰为参照峰,计算各特征峰的相对保留时间及相对峰面积,按平均数法(或中位数法)生成对照图谱,建立地榆药材对照特征图谱(如图3所示);标识将各供试品的共有峰(参见图4);选择与地榆对照药材保留时间一致的4个共有峰,作为特征峰,分别第一峰(1号峰,参见图4中1(s))、第二峰(2号峰,参见图4中2)、第三峰(3号峰,参见图4中3)和第四峰(4号峰,参见图4中4)。1.5.2特征图谱测定结果对23批地榆药材特征图谱进行分析,以没食子酸色谱峰为参照峰s,计算各特征峰与s峰的相对保留时间和相对峰面积,并计算rsd值,实验结果见表2、表3。表223批地榆药材特征图谱(相对保留时间)表323批地榆药材特征图谱(相对峰面积)1.6含量测定1.6.3含量测定结果取地榆药材样品,按照“1.3”项下制备供试品溶液,按"1.1"项下色谱条件进行测定分析,采用外标法计算地榆药材中没食子酸、鞣质的含量(见表4)。鞣质的含量测定参照2020版《中国药典》中照鞣质含量测定法(通则2202)测定,结果如下:表423批地榆药材含量测定结果结果分析与讨论:结果显示,23批地榆饮片的没食子酸含量(1.6%~3.8%),鞣质含量(8.9%~23.6%)均符合《中国药典》2020年版地榆饮片项下的含量要求(没食子酸不得少于0.6%,鞣质不得少于2.0%),且与对应原药材含量相近。2uplc-qtof/ms方法与结果2.1液相-质谱条件色谱柱:watersacquityuplcbehc18(2.1mm×100mm,1.7μm);以甲醇为流动相a,以0.1%甲酸溶液为流动相b,按表5中的规定进行梯度洗脱;流速为每分钟0.25ml;柱温为35℃;检测波长为270nm。质谱离子源与扫描参数设置详细见表6。表5梯度洗脱表表6质谱参数表2.2供试品的制备供试品的制备同“1.3”项下。2.3指认结果2.3.1采用上述液相色谱和质谱分析条件,对供试品溶液进行检测,得到相应的质谱图与紫外吸收图,见图5。(1)1号峰提取1号峰的一级色谱图,时间段为4.49-4.67min。结果见图6-图7。从图6和图7可以看出,在1号峰的一级色谱提取图中,正离子模式下m/z171.0288峰的响应值最高,为[m+h]+峰;负离子模式下有m/z169.0132峰,为[m-h]-峰;从其精确分子量推测其分子式可能为c9h11no2;提取m/z171.0288峰碰撞能量为40的二级图,结果显示,主要碎片离子有153.0183、127.0391、125.0235、109.0288、107.0132、81.0341,详细结果见图8。提取m/z169.0132峰碰撞能量为40的二级图,结果显示,主要离子碎片有125.0232等,详细结果见图9。将该信号峰的精确分子量及二级离子碎片信息与本地质谱数据库中的化合物光谱进行匹配,结果显示,m/z171.0288和m/z169.0132峰匹配度最高的化合物为没食子酸,匹配度为94.62%,二级碎片匹配情况见图10,没食子酸结构式见图11。没食子酸可能的裂解方式如图12-图13所示,根据碎片离子对比结果和结构式的裂解推断分析,提示该化合物可能为没食子酸。(2)2号峰提取2号峰的一级色谱图,时间段为13.28-13.50min。结果见图14-图15。从图14-图15可以看出,在2号峰的一级色谱提取图中,正离子模式下有m/z315.0710峰,负离子模式下有m/z345.0829峰;提取正离子模式下有m/z315.0710峰碰撞能量为40的二级质谱图,结果显示,主要离子碎片有153.0182等,详细结果见图16。提取负离子模式下有m/z345.0829峰碰撞能量为40的二级质谱图,结果显示,主要离子碎片有239.0594、148.0155、223.0280、87.0438等,详细结果见图17。(3)3号峰提取3号峰的一级色谱图,时间段为17.53-17.70min。结果见图18-图19。从图18-图19可以看出,在3号峰的一级色谱提取图中,正离子模式下有m/z291.0861峰,为[m+h]+峰,提取m/z291.0861峰碰撞能量为40的二级质谱图,结果显示,主要离子碎片有139.0390、123.0442、147.0441等,结果见图20;负离子模式下有m/z289.0720峰,为[m-h]-峰,提取m/z289.0720峰碰撞能量为40的二级质谱图,结果显示,主要离子碎片有109.0282、125.0232、123.0440、151.0390等,结果见图21。从其精确地分子量推测其分子式可能为c15h14o6。将该信号峰的精确分子量及二级离子碎片信息与本地质谱数据库中的化合物光谱进行匹配,结果显示,m/z291.0861与m/z289.0720峰匹配度最高的化合物为表儿茶素或儿茶素(同分异构体),匹配度为94.24%,二级碎片匹配情况见图22-图23,儿茶素结构式见图24,可能的裂解碎片见图25-图26。根据碎片离子对比结果和结构式的裂解推断分析,提示该化合物可能为儿茶素或表儿茶素(同分异构体)。(4)4号峰提取4号峰的一级色谱图,时间段为30.37-30.71min。结果见图27。从图27可以看出,在6号峰的一级色谱提取图中,负离子模式下有m/z300.9991峰,为[m-h]-峰,提取m/z300.9991峰碰撞能量为40的二级质谱图,结果显示,主要离子碎片有300.9991,结果见图28。从其精确地分子量推测其分子式可能为c14h6o8。将该信号峰的精确分子量及二级离子碎片信息与本地质谱数据库中的化合物光谱进行匹配,结果显示,m/z300.9991峰匹配度最高的化合物为鞣花酸,匹配度为89.24%,二级碎片匹配情况见图29,鞣花酸结构式见图30。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1