一种手性镇痛类多肽药物的纯化方法与流程
1.本发明涉及药物分离纯化技术领域,尤其涉及一种手性镇痛类多肽药物的纯化方法。
背景技术:
2.阿片样物质受体是一类主要的g蛋白偶联受体,是内源性阿片肽以及阿片类药物结合靶点,广泛存在于中枢神经系统和外周神经系统。阿片受体激活后对神经系统免疫以及内分泌系统具有调节作用,是目前最强且常用的中枢镇痛药。内源性阿片肽是哺乳动物体内天然生成的阿片样活性物质,目前已知的内源性阿片肽大致分为脑啡肽、内啡肽、强啡肽和新啡肽几类。中枢神经系统中存在其相应的阿片受体,即μ、δ和κ受体。μ受体镇痛活性最强,成瘾性也最强,是产生副作用的主要原因。δ受体成瘾性小,镇痛作用也不明显。κ受体(kor)镇痛活性介于前两者之间。多肽类kor激动剂能在不进入中枢的情况下在外周发挥镇痛作用,不会导致呼吸抑制和便秘等毒副作用,且成瘾性更低,因此具有药物成瘾治疗的潜力。
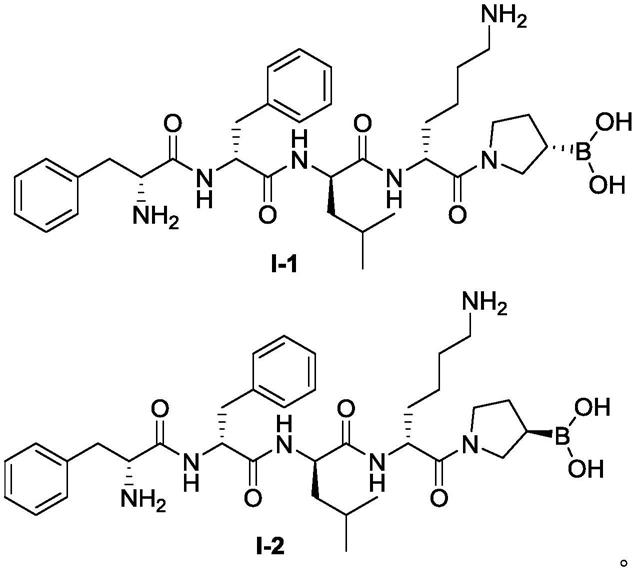
3.专利cn111233974b报道了一系列具有优异的激动活性的新型kor激动剂,包括结构如式i
‑
1所示的化合物(化学名:[(s)
‑1‑
(d
‑
苯丙氨酰
‑
d
‑
苯丙氨酰
‑
d
‑
亮氨酰
‑
d
‑
赖氨酰)吡咯烷
‑3‑
基]硼酸盐酸盐)。化合物i
‑
1的粗品中含有多种工艺杂质、降解杂质以及最难分离的手性异构体杂质(式i
‑
2,化学名:[(r)
‑1‑
(d
‑
苯丙氨酰
‑
d
‑
苯丙氨酰
‑
d
‑
亮氨酰
‑
d
‑
赖氨酰)吡咯烷
‑3‑
基]硼酸盐酸盐),这些异构体杂质与主成分的极性差异特别小,理化性质极其类似,给检测分析、分离纯化带来了极大的挑战,能否有效获得符合临床所需以及药物研究所需质量的样品是对分离纯化的巨大挑战。因此,开发一种简便高效、收率良好的纯化工艺解决产品质量、收率、成本以及操作便利性等问题顺利保障创新药物开发的临床以及后续所需极为迫切。
[0004]
技术实现要素:
[0005]
有鉴于此,本发明要解决的技术问题在于提供一种手性镇痛类多肽药物i
‑
1的纯化方法,具有较高的收率和纯度。
[0006]
为达到上述目的,本发明提供了一种手性镇痛类多肽药物i
‑
1的纯化方法,包括以下步骤:
[0007]
s1)将手性镇痛类多肽药物粗品与甲醇混合溶解,然后加入tfa水溶液稀释,过滤,得到手性镇痛类多肽药物初纯溶液;
[0008]
s2)将手性镇痛类多肽药物初纯溶液通过反相色谱法纯化后获得手性镇痛类多肽药物精纯溶液;所述反相色谱法中,采用十八烷基硅烷键合硅胶为固定相,甲酸水溶液为a相,甲醇为b相;进行梯度洗脱;
[0009]
s3)将手性镇痛类多肽药物精纯溶液通过hplc法转盐,得到镇痛类多肽药物盐溶液;所述盐溶液经减压浓缩、干燥后得到镇痛类多肽药物纯品。
[0010]
本领域技术人员公知:多肽的结构差异极大,手性较多,工艺杂质与各种降解杂质均极难控制,虽然在分析检测中有可能可以有效分离相关杂质,但分析检测由于其色谱条件苛刻,上样量极小,色谱填料粒径极细,成本极高,设备仪器相应要求极高,故其分析方法几乎不可能在工业级纯化制备中实现放大生产。本发明通过大量的纯化实验摸索开发了一种独属于该手性镇痛类多肽药物的纯化制备工艺,解决了工业纯化制备的瓶颈与难点。
[0011]
本发明采用反相色谱纯化和转盐,解决了手性杂质对产物的质量影响,解决了镇痛类多肽药物高温降解和氧化降解的几率。
[0012]
本发明中,所述手性镇痛类多肽药物粗品由液相合成法或固相合成法合成。
[0013]
本发明中,所述手性镇痛类多肽药物粗品中包括其手性杂质。
[0014]
在本发明的一些具体实施例中,所述手性镇痛类多肽药物粗品包括[(s)
‑1‑
(d
‑
苯
丙氨酰
‑
d
‑
苯丙氨酰
‑
d
‑
亮氨酰
‑
d
‑
赖氨酰)吡咯烷
‑3‑
基]硼酸盐酸盐(式i
‑
1所示)和[(r)
‑1‑
(d
‑
苯丙氨酰
‑
d
‑
苯丙氨酰
‑
d
‑
亮氨酰
‑
d
‑
赖氨酰)吡咯烷
‑3‑
基]硼酸盐酸盐(式i
‑
2所示)。
[0015]
首先,所述手性镇痛类多肽药物粗品加入甲醇中溶解。
[0016]
本发明优选的,所述手性镇痛类多肽药物粗品与甲醇的质量体积比为1:9~12(g:ml)。
[0017]
然后加入tfa水溶液稀释,过滤,得到手性镇痛类多肽药物初纯溶液。
[0018]
本发明优选的,所述tfa水溶液的浓度为0.1%~0.3%,更优选为0.3%。
[0019]
本发明优选的,所述tfa水溶液与手性镇痛类多肽药物粗品的体积质量比为1~1.5:2(ml:g)。
[0020]
本发明优选的,加入tfa水溶液稀释后,所述手性镇痛类多肽药物在溶液中的浓度为3mg/ml~9mg/ml,更优选为6mg/ml。
[0021]
然后进行反相色谱法纯化。
[0022]
所述反相色谱法采用十八烷基硅烷键合硅胶为固定相。
[0023]
所述十八烷基硅烷键合硅胶的粒径优选为10um,孔径优选为
[0024]
所述流动相a相优选为甲酸水溶液或乙酸水溶液。
[0025]
所述甲酸水溶液或乙酸水溶液的浓度优选为0.1%~0.2%。
[0026]
所述流动相b相优选为甲醇或乙腈,进一步优选为甲醇。
[0027]
本发明优选的,所述步骤s2)的梯度洗脱具体为:
[0028]
b相在5min内维持5%梯度,b相由5.1min至50min由5%升至55%梯度,并维持b相55%梯度至少10min,所述a相为0.15%甲酸水溶液。
[0029]
上述反相色谱法主要解决了非对映异构体杂质,且能够有效控制其他工艺杂质,同时对极易出现的水解、氧化杂质进行了有效控制。
[0030]
本发明优选的,所述hplc法转盐具体为:
[0031]
使用无机盐和强酸组成的缓冲体系通过反相色谱交换转盐。
[0032]
所述无机盐优选为氯化铵或其他氯化物,如氯化钠、氯化钾等。
[0033]
所述强酸优选为盐酸。
[0034]
进一步优选的,所述hplc法中,流动相a1相为氯化铵水溶液(盐酸调节ph值为3.5),a2相为盐酸水溶液,进一步优选为0.005%~0.01%的盐酸水溶液,所述b相为乙腈或甲醇,进一步优选为甲醇。
[0035]
所述氯化铵水溶液的浓度优选为20mm。
[0036]
本发明优选的,所述hplc法转盐的梯度设置为,a1相在60min内维持95%梯度,a2相由60.1min至80min维持95%梯度,80.1min至120min内b相由5%升至60%梯度且至少保持b相60%的梯度30min,所述b相为甲醇。
[0037]
进一步优选的,所述纯化梯度条件的线速度流速为5.5~6.5cm/min,转盐梯度条件的线速度流速为3.5~4.5cm/min。
[0038]
本发明优选的,所述步骤s3)中减压浓缩的温度为小于30℃,真空度为0.08以上。
[0039]
本发明优选的,所述浓缩过程避免光线直射。
[0040]
本发明优选的,所述步骤s3)中的干燥为减压冷冻干燥。
[0041]
所述减压冷冻干燥的过程优选为:
[0042]
预冻至
‑
50~
‑
10℃保持2~4小时,一阶干燥
‑
5℃干燥36~48小时,升华干燥20℃,干燥时间36~48h,真空度恒定在0.01~1.00mba。
[0043]
与现有技术相比,本发明提供了一种手性镇痛类多肽药物i
‑
1的纯化方法,包括以下步骤:s1)将手性镇痛类多肽药物粗品与甲醇混合溶解,然后加入tfa水溶液稀释,过滤,得到手性镇痛类多肽药物初纯溶液;s2)将手性镇痛类多肽药物初纯溶液通过反相色谱法纯化后获得手性镇痛类多肽药物精纯溶液;所述反相色谱法中,采用十八烷基硅烷键合硅胶为固定相,甲酸水溶液或乙酸水溶液为a相,甲醇或乙腈为b相;进行梯度洗脱;s3)将手性镇痛类多肽药物精纯溶液通过hplc法转盐,得到镇痛类多肽药物盐溶液;所述盐溶液经减压浓缩、干燥后得到镇痛类多肽药物i
‑
1纯品。
[0044]
本发明通过工艺步骤和参数的合理优化,使非对映异构体和其他工艺以及降解杂质均不大于0.5%,总杂不大于1.0%,且使纯度大于99%时其收率也达到50%以上,开创性的解决了制备高纯度的手性镇痛类多肽药物i
‑
1的纯化技术难点。
附图说明
[0045]
图1为纯化前样品(粗品)检测图谱;
[0046]
图2为实施例1中纯化后样品纯度检测图谱;
[0047]
图3为实施例2中纯化后样品纯度检测图谱;
[0048]
图4为实施例3中纯化后样品纯度检测图谱;
[0049]
图5为实施例3中纯化样品浓缩后纯度检测图谱;
[0050]
图6为实施例4中纯化后样品纯度检测图谱。
具体实施方式
[0051]
为了进一步说明本发明,下面结合实施例对本发明提供的手性镇痛类多肽药物i
‑
1的纯化方法进行详细描述。
[0052]
以下实施例中镇痛类多肽药物i
‑
1粗品按照专利cn111233974b实施例2公开的方法制备得到。
[0053]
实施例1
[0054]
一种纯化制备手性镇痛类多肽药物i
‑
1的方法,包括:
[0055]
(1)样品处理:采用固相合成法获得粗品纯度达54.67%的镇痛类多肽药物i
‑
1粗品,用甲醇按照120g/l的浓度溶解,搅拌,完全溶解后用0.3%的tfa水溶液稀释至约6mg/ml,再用0.45um的有机系滤膜过滤,滤除不溶物,得到粗品滤液。
[0056]
(2)纯化:将上述粗品溶液约2000ml,采用dac150液相纯化系统,填料为fuji c18,10um,装填高度250mm。流动相中a相为:0.15%甲酸水溶液;流动相b相为:甲醇;体积流速:1100ml/min;梯度:5min内b相5%~5%,5.1min至50min间b相由5%升至55%梯度,并维持b相55%梯度至少10min,检测波长:220nm;分段收集主峰,合并任意单一杂质不大于0.5%的分段纯化液,不合格段合并浓缩后重复上述纯化步骤至纯化液合格。
[0057]
(3)转盐
[0058]
反相色谱条件:以十八烷基硅烷键合硅胶为固定相的高效液相色谱制备柱,柱子的粒径10um,孔径100a。柱子直径和长度为:150x250mm。流动相中a1相为:20mm氯化铵,通过
盐酸调整ph至3.5;流动相中a2相为:0.005%盐酸水溶液;流动相中b相为:甲醇;体积流速:700ml/min;a1相在60min内维持95%梯度,切换a2相,a2相由60.1min至80min维持95%梯度,80.1min至120min内b相由5%升至60%梯度且至少保持b相60%的梯度30min,所述b相为甲醇。
[0059]
将收集的有关物质复核要求的镇痛类多肽药物于水温30℃以下的条件下减压旋蒸浓缩至浓度约为20mg/ml~40mg/ml,然后冷冻干燥获得任意单一杂质不大于0.5%,总杂不大于1.0%,纯度高于99.0%的手性镇痛类多肽药物溶液。
[0060]
经过计算:本实施例的最大单一杂质为手性非对映异构体杂质0.17%,总杂为0.31%,纯度为99.69%,纯化收率为58.2%。
[0061]
实施例2
[0062]
(1)样品处理:采用固相合成法获得粗品纯度达58.94%的镇痛类多肽药物i
‑
1粗品,用甲醇按照120g/l的浓度溶解,搅拌,完全溶解后用0.3%的tfa水溶液稀释至约6mg/ml,再用0.45um的有机系滤膜过滤,滤除不溶物,得到粗品滤液。
[0063]
(2)纯化:将上述粗品溶液约2000ml,采用dac150液相纯化系统,填料为nano unisil c18,10um,装填高度250mm。流动相中a相为:0.10%甲酸水溶液;流动相b相为:甲醇;体积流速:1000ml/min;梯度:5min内b相5%~5%,5.1min至50min间b相由5%升至55%梯度,并维持b相55%梯度至少10min,检测波长:220nm;分段收集主峰,合并任意单一杂质不大于0.5%的分段纯化液,不合格段合并浓缩后重复上述纯化步骤至纯化液合格。
[0064]
(3)转盐
[0065]
色谱柱:以十八烷基硅烷键合硅胶为固定相,柱子直径和长度为150mm
×
250mm
[0066]
流动相:a1:20mm氯化铵酸水溶液,a2相0.01%盐酸水溶液,b:甲醇
[0067]
检测波长:220nm
[0068]
体积流速1:750ml/min,体积流速2:1100ml/min
[0069]
转盐洗脱梯度如表1所示:
[0070]
表1
[0071]
[0072]
将收集的有关物质符合要求的镇痛类多肽药物于水温30℃以下的条件下减压旋蒸浓缩至浓度约为20mg/ml~40mg/ml,然后冷冻干燥获得任意单一杂质不大于0.5%,总杂不大于1.0%,纯度高于99.0%的手性镇痛类多肽药物溶液。
[0073]
经过计算:本实施例的最大单一杂质为手性非对映异构体杂质0.18%,总杂为0.38%,纯度为99.62%,纯化收率为61.3%。
[0074]
对比例1
[0075]
采用实施例1中的反相色谱进行纯化,转盐过程条件有所变化,具体过程和实验结果如下:
[0076]
一种纯化制备手性镇痛类多肽药物i
‑
1的方法,包括:
[0077]
(1)样品处理:采用固相合成法获得粗品纯度达55.34%的镇痛类多肽药物粗品,用甲醇按照120g/l的浓度溶解,搅拌,完全溶解后用0.3%的tfa水溶液稀释至约6mg/ml,再用0.45um的有机系滤膜过滤,滤除不溶物,得到粗品滤液。
[0078]
(2)纯化:将上述粗品溶液约2000ml,采用dac150液相纯化系统,填料为fuji c18,10um,装填高度250mm。流动相中a相为:0.15%甲酸水溶液;流动相b相为:甲醇;体积流速:1100ml/min;梯度:5min内b相5%~5%,5.1min至50min间b相由5%升至55%梯度,并维持b相55%梯度至少10min,检测波长:220nm;分段收集主峰,合并任意单一杂质不大于0.5%的分段纯化液,不合格段合并浓缩后重复上述纯化步骤至纯化液合格。
[0079]
(3)转盐
[0080]
色谱柱:以十八烷基硅烷键合硅胶为固定相,柱子直径和长度为150mm
×
250mm
[0081]
流动相:a1:30mm氯化铵酸水溶液,a2相0.03%盐酸水溶液,b:甲醇
[0082]
检测波长:220nm
[0083]
体积流速1:750ml/min,体积流速2:1000ml/min
[0084]
转盐洗脱梯度如表2所示:
[0085]
表2
[0086][0087][0088]
将收集的有关物质符合要求的镇痛类多肽药物于水温30℃以下的条件下减压旋
蒸浓缩至浓度约为20mg/ml~40mg/ml,然后冷冻干燥获得手性镇痛类多肽药物成品。
[0089]
由于本品转盐条件发生变化,转盐中的盐酸含量增加导致合格纯化液浓缩过程中盐酸浓度不断增加,合格纯化液在浓缩过程中其降解杂质(酸水解杂质)在急剧增加,由纯化后浓缩前的0%增加至浓缩后的2.41%。
[0090]
经过计算:本对比例最终产品的最大单一杂质为酸水解杂质为2.41%,总杂为2.55%,纯度为97.44%,纯化收率为48.52%。
[0091]
实施例3
[0092]
(1)样品处理:采用固相合成法获得粗品纯度达55.8%的镇痛类多肽药物i
‑
1粗品,用甲醇按照120g/l的浓度溶解,搅拌,完全溶解后用0.3%的tfa水溶液稀释至约6mg/ml,再用0.45um的有机系滤膜过滤,滤除不溶物,得到粗品滤液。
[0093]
(2)纯化:将上述粗品溶液约2000ml,采用dac150液相纯化系统,填料为nano unisil c18,10um,装填高度250mm。流动相中a相为:0.15%甲酸水溶液;流动相b相为:甲醇;体积流速:1100ml/min;梯度:5min内b相5%~5%,5.1min至50min间b相由5%升至55%梯度,并维持b相55%梯度至少10min,检测波长:220nm;分段收集主峰,合并任意单一杂质不大于0.5%的分段纯化液,不合格段合并浓缩后重复上述纯化步骤至纯化液合格。
[0094]
(3)转盐
[0095]
反相色谱条件:以十八烷基硅烷键合硅胶为固定相的高效液相色谱制备柱(填料为nano unisil c18,10um,直径150mm,装填高度250mm),流动相中a1相为:20mm氯化铵,通过盐酸调整ph至3.5;流动相中a2相为:0.002%盐酸水溶液;流动相中b相为:甲醇;体积流速:750ml/min;a1相在60min内维持95%梯度,切换a2相,同时流速提升至1100ml/min,a2相由60.1min至80min维持95%梯度,80.1min至120min内b相由5%升至60%梯度且至少保持b相60%的梯度30min,所述b相为甲醇。
[0096]
将收集的有关物质符合要求的镇痛类多肽药物于水温30℃以下的条件下减压旋蒸浓缩至浓度约为20mg/ml~40mg/ml,然后冷冻干燥获得手性镇痛类多肽药物i
‑
1成品。
[0097]
经过计算:本实施例的最大单一杂质为手性非对映异构体杂质0%,总杂为0.42%,纯度为99.58%,纯化收率为69.78%。
[0098]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1