蒽醌类化合物的提取方法
1.本发明属于化合物提取分析技术领域,具体涉及一种蒽醌类化合物的提取方法,是一种基于超声辅助功能化离子液体分散液液微萃取提取分析中草药中蒽醌类化合物的方法。
背景技术:
2.近年来决明子、番泻叶、芦荟、大黄等中药被广泛应用于保健食品中,蒽醌类成分是这些中药的重要功效成分。美国食品药品监督管理局(fda)番泻叶列为非处方药,其中主要的活性成分就是蒽醌类物质。蒽醌类物质按母核可分为单核蒽醌和双核蒽醌,其中单核蒽醌主要包括大黄酸、芦荟大黄素、大黄酚、大黄素等。其中,美国国家毒理学计划对大黄素进行的毒理学和致癌性研究结果表明,暴露于大黄素会导致致癌风险和发病率增加,验证了蒽醌类物质可能有致癌风险的说法。2011年,欧洲实验室首次对于来自中国、印度和斯里兰卡的进口红茶中的蒽醌类物质进行检测,欧盟规定茶叶的最大残留限量为0.02mg/kg欧洲。出于安全考虑,有必要对此类中草药的蒽醌类化合物进行提取和定量分析,为此类中草药的安全性评价提供数据支持。
3.目前针对富含单核蒽醌的中药,如大黄,通常采用的提取方法为先使用乙醚回流提取蒽醌类化合物后再使用有机溶剂液液萃取,但是使用该方法有明显的缺点,包括目标分析物的损失、检测限高、通常还需要另外进行浓缩处理等。特别是,回流提取需要使用大量溶剂,而这些溶剂通常是危险的,会导致产生大量的有毒废液。基于此,需要建立一种绿色、环保、准确、有效、省时的替代回流提取的方法,从而减少有毒溶剂的消耗,节省时间和成本。
4.中药材中化合物测定过程中最重要的步骤是样品的前处理。经过样品的前处理,一方面可以起到浓缩待测痕量组分的消除干扰的作用,从而提高方法的灵敏度,降低方法的检出限;另一方面可以去除对仪器或管道有害的物质,从而延长仪器的使用寿命。而分散液液微萃取(dllme)是一种新型的的微萃取技术。当样品溶液中加入萃取剂和分散剂,分散剂使萃取剂均匀分布于样品溶液中,增大了两者之间的接触面积,从而使待测物被萃取剂快速萃取,在离心之后留在离心管底部,形成一定体积的沉淀相。分散液液微萃取法较传统的液液萃取法来说,集采样、萃取和浓缩于一体,具有有机溶剂使用量少、萃取效率高、富集倍数大、操作方便简单、环境友好等特点,是一种绿色的样品前处理方法,在痕量分析领域具有广阔的前景。
5.而苄基功能化离子液体具有独特的理化性质,引入苄基使得该离子液体疏水性升高,更有利于对蒽醌类化合物的萃取,可作为有机溶剂的替代品,使其成为一种良好的萃取溶剂。离子液体阴离子可能与目标化合物的酚羟基相互作用,尤其是氢键相互作用,有利于目标化合物的溶解和萃取,与传统溶剂萃取相比提高了萃取率。基于此,就萃取技术而言,苄基功能化咪唑盐离子液体可能具有更好的技术优势,但目前还没有将此类离子运用于蒽醌类化合物中的报道,也缺乏一种利用此类离子液体进行的微萃取技术。
技术实现要素:
6.本发明旨在解决上述问题,提供了一种蒽醌类化合物的提取方法,采用了疏水性溶剂超声辅助分散液
‑
液微萃取
‑
高效液相色谱法,操作简单、检测速度快、成本低廉且绿色环保。
7.按照本发明的技术方案,所述蒽醌类化合物的提取方法,蒽醌类化合物的结构式如下:
[0008][0009]
其中,r1为h或oh,r2为ch2oh、cooh或ch3,包括以下步骤,
[0010]
s1:调节待萃取基质的ph为1
‑
7;
[0011]
s2:加入萃取剂,得到混合液体,所述萃取剂为苄基功能化离子液体溶液;
[0012]
s3:将所述混合液体进行超声处理;
[0013]
s4:将超声处理后的混合液体冷却后离心,获得沉降物,完成蒽醌类化合物的提取。
[0014]
具体的,蒽醌类化合物为大黄素(emodin)、大黄酚(chrysophanol)、大黄酸(rhein)和芦荟大黄素(aloe
‑
emodin),其结构式如下:
[0015][0016]
进一步的,所述步骤s1中,待萃取基质的制备方法如下:将样品研磨后,加入水煮沸提取1
‑
3h;冷冻干燥,取冻干粉加入水,超声提取0.4
‑
1h;离心,所得上清液即为待萃取基质。
[0017]
进一步的,每5ml待萃取基质,加入萃取剂中的苄基功能化离子液体的体积为30
‑
100μl,优选的,加入体积为50μl。
[0018]
进一步的,所述苄基功能化离子液体为苄基咪唑亚胺盐离子液体。
[0019]
具体的,所述苄基咪唑亚胺盐离子液体为1
‑
苄基
‑3‑
甲基咪唑双(三氟甲烷黄酰)亚胺盐。
[0020]
进一步的,所述苄基功能化离子液体溶液的溶剂为乙腈、甲醇和丙酮中的一种或多种,苄基功能化离子液体溶液中溶质与溶剂的体积比为1:8
‑
20,优选为1:14。
[0021]
进一步的,所述步骤s3中,超声处理的温度为10
‑
50℃,优选为20℃;时间为0.1
‑
12min,优选为6min。
[0022]
具体的,步骤s3中,ph优选为4;步骤s3中,将所述混合液体涡旋混合后,再进行超声处理;步骤s4中,冷却为冰浴冷却,冷却时间为8
‑
15min(优选为10min)。
[0023]
基于上述提取方法,可以提供了一种蒽醌类化合物的检测方法,通过高效液相色谱仪的dad检测器,对上述提取方法获得的沉降物进行检测。
[0024]
本发明以疏水性离子液体作为超声辅助分散液液微萃取的萃取剂对待测样品中的四种蒽醌物质进行萃取分离,再通过高效液相色谱法对四种蒽醌物质进行分析检测,所述疏水性离子液体为1
‑
苄基
‑3‑
甲基咪唑双(三氟甲烷黄酰)亚胺盐([bemim][tf2n]),该离子液体(il)阴离子可能与目标化合物的羟基相互作用,尤其是氢键相互作用有利于目标化合物的溶解和萃取,[bemim][tf2n]与蒽醌类化合物的酚羟基之间的相互作用,使其相较与其他离子液体而言,大大提高了萃取效率,回收率提高,富集倍数增加,与传统溶剂萃取相比提高了萃取率。在超声波辅助的条件下在试管内形成稳定的混浊溶液,帮助萃取溶剂的细小液滴的形成,提高萃取效率。
[0025]
本发明的另一方面提供了苄基功能化离子液体在蒽醌类化合物微萃取中的应用。
[0026]
进一步的,所述苄基功能化离子液体为苄基咪唑亚胺盐离子液体。具体的,所述苄基咪唑亚胺盐离子液体为1
‑
苄基
‑3‑
甲基咪唑双(三氟甲烷黄酰)亚胺盐。
[0027]
本发明的技术方案相比现有技术具有以下优点:
[0028]
1)使用本方法测定蒽醌类物质(特别是中草药中的四种蒽醌类物质)灵敏度高,检出限低,在0.4
‑
0.8ng/ml。
[0029]
2)本方法所使用的萃取溶剂少(每5ml待萃取基质,加入离子液体30
‑
100μl,甲醇分散剂0.7ml左右),相比于传统的回流提取提取方法,节省了大量的对人体有害的有机溶剂。
[0030]
3)本方法样品的前处理时间段(<30min),与传统方法相比,节省了分析时间,因此可以用于大批次的蒽醌类化合物的测定。
附图说明
[0031]
图1为芦荟大黄素、大黄酸、大黄素、大黄酚混合标准品溶液hplc图。
[0032]
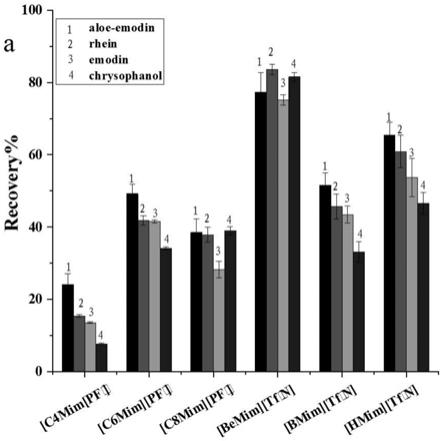
图2为不同离子液体充当萃取剂对蒽醌类化合物萃取效果的影响。
[0033]
图3为不同[bemim][tf2n]体积对萃取结果的影响。
[0034]
图4为不同提取温度对萃取结果的影响。
[0035]
图5为不同超声时间对萃取结果的影响。
具体实施方式
[0036]
下面结合附图和具体实施例对本发明作进一步说明,以使本领域的技术人员可以更好地理解本发明并能予以实施,但所举实施例不作为对本发明的限定。
[0037]
实施例1蒽醌类化合物提取方法中控制参数的确定
[0038]
影响超声辅助萃取和分散液液微萃取效率的主要因素包括:萃取剂类型和用量、超声时间、提取温度、分散剂类型和用量,以及离子强度和ph。
[0039]
1、超声辅助萃取剂离子种类的确定:离子液体在微萃取过程中充当萃取剂。由于不同的离子液体对目标分析物的萃取能力有不同的影响,因此离子液体的选择对最终的萃
取结果非常重要。本发明选择了三种基于[tf2n]阴离子的离子液体和三种基于[pf6]阴离子的不同阳离子用于中草药提取物中蒽醌的富集和提取,其中,三种基于[tf2n]阴离子的离子液体分别为1
‑
丁基
‑3‑
甲基咪唑双(三氟甲烷磺酰)亚胺盐([bmin][tf2n])、1
‑
己基
‑3‑
甲基咪唑双(三氟甲烷磺酰)亚胺盐([hmin][tf2n])和1
‑
苄基
‑3‑
甲基咪唑双(三氟甲烷磺酰)亚胺盐([bemin][tf2n]),三种基于[pf6]阴离子的不同阳离子分别为1
‑
丁基
‑3‑
甲基咪唑六氟磷酸盐([c4mim[pf6])、1
‑
己基
‑3‑
甲基咪唑六氟磷酸盐([c6mim[pf6])、1
‑
辛基
‑3‑
甲基咪唑六氟磷酸盐([c8mim[pf6]。由于[tf2n]阴离子的电荷分布更均匀,对称性更低,因此具有很强的疏水性、耐水性和较低的粘度,其次[tf2n]阴离子能与蒽醌类化合物形成特殊氢键,这将促进萃取。对于具有良好回收率的[tf2n]
‑
阴离子型离子液体,[bemim][tf2n]的回收率最高,因为引入的苄基使得其疏水性更强,回收率更好,达到81%(如图2所示),因此发明选择[bemim][tf2n]作为dllme过程中的萃取剂。
[0040]
2、[bemim][tf2n]体积的确定:离子液体的用量是影响萃取效率的关键因素,萃取剂的用量直接影响回收率的水平。本发明选择30
‑
100μl[bemim][tf2n]。实验结果表明(如图3所示),在实验中使用50μl[bemim][tf2n]获得令人满意的富集因子和提取回收率。
[0041]
3、提取温度的确定:在萃取过程中,一般提高温度可以加速分析物在两相之间的转移,也可以增加萃取剂和目标分析物在溶液中的溶解度。在发明中,在5
‑
50℃下研究了温度对提取效率的影响。其中在20℃范围内获得最佳回收率(如图4所示)。
[0042]
4、超声时间的确定:在20℃下进行超声辅助提取,在0
‑
10min范围内评价超声处理时间的效果(如图5所示)。最终实验选择超声辅助提取6min,此时提取效果最佳。
[0043]
5、分散剂和体积的选择:本研究选择了三种有机溶剂,乙腈、甲醇和丙酮,它们在离子液体和水相中都具有适当的混溶性。四种目标分析物的回收率相似,但考虑到hplc中使用甲醇作为流动相,因此选择甲醇作为分散剂。分散剂的体积会影响萃取剂在水样中的分散效果。本实验中固定萃取剂用量为50μl,优化了0.4
‑
1.0ml不同体积的甲醇,其中甲醇体积为0.7ml提取效果最佳。
[0044]
6、ph的选择:样品溶液的ph值决定了分析物的存在形式(离子或中性形式),此后可能会影响提取效率。通过向样品中加入适当的磷酸或氢氧化钠用以调节ph,在1
‑
7范围内评估样品ph值的影响。当样品ph等于4时,芦荟大黄素、大黄酸和大黄素的回收率最高。因此,在以下研究中选择了ph4。
[0045]
7、离子强度选择:离子强度可以提高目标分析物在萃取剂中的溶解度,从而提高萃取效率。在本研究中,将不同量的nah2po4(从0%到20%)添加到样品中以评估离子强度的影响。加入nah2po4后,萃取回收率下降。因此,在本发明实验中不添加na
+
盐。
[0046]
实施例2番泻叶样品中蒽醌类化合物的测定
[0047]
1)标准溶液的配制:单一标准品储备液:精密称取大黄酚、大黄素、芦荟大黄素、大黄酸各2.15mg、1.25mg、1.30mg、1.40mg,加50%甲醇(甲醇与水体积比为1:1)溶解,分别定容至10ml,配置成浓度分别为215μg/ml、125μg/ml、130μg/ml、140μg/ml的单一标准品储备液。混合标准品溶液:精密吸取各单一标准品储备液适量,配置成相应浓度梯度的混合标准品溶液(混合标准品溶液hplc图如图1所示)。
[0048]
2)样品处理:将番泻叶样品研磨成细粉,取番泻叶细粉100g置于3000ml不锈钢锅中,加入1500ml蒸馏水,煮沸提取1.5h后,取200ml提取液冻干进行保存。将0.5g上述冻干粉
加入20ml蒸馏水,超声提取30min后。将混合溶液离心5min以分离冻干粉末残留物和水溶液。
[0049]
3)离子液体分散液液微萃取(il
‑
dllme):将5ml的上述提取液置于10ml锥形底离心管中。使用注射器将50μl[bemim][tf2n](已溶解在700μl甲醇中)快速注入样品溶液中。对于产生混浊样品溶液,涡旋3分钟,在20℃的条件下超声6min后冰浴10min,最后在3500rmp条件下离心5分钟。丢弃上层水相,量取在离心管底部il相,通常为26μl,小心地取出注入hplc系统。
[0050]
4)hplc测定:色谱分析是在配有二极管阵列检测(dad)系统的chromaster型hplc系统上进行的。反相c18色谱柱(内径250mm
×
4.6mm,5μm)型号为agilenteclipsexdb
‑
c
18
。流动相由水(a)添加0.05%磷酸和甲醇(b)(比例为20:80(v/v))组成,流速为1.0ml
·
min
‑1。进样量和检测波长分别为10μl和254nm。
[0051]
5)方法性能评估:为了研究所提出的测定样品中蒽醌类化合物的方法的方法性能,设计了一系列实验来评估优化条件下方法的线性、重现性、检测限和其他特性等参数。具体结果见表1。在芦荟大黄素、大黄酚、大黄酸、大黄素分析物均表现出良好的线性,相关系数(r2)范围为0.9996至0.9999。该方法的精密度是通过对加标样进行五次萃取和分析来确定的,相对标准偏差(rsd)范围为1.25%至5.28%。根据信噪比(s/n=3),检测限(lod)的计算范围为0.50
‑
2.02μg
·
l
‑1。这些参数表明,本方法具有高灵敏度和可靠性,可用于检测样品中蒽醌类化合物的浓度。
[0052]
表1方法性能评估
[0053][0054]
6)样品测定:为验证所提方法在实际样品分析中的适用性,对采自印度三个不同地区(ⅰ、ⅱ、ⅲ)的番泻叶样品中的蒽醌类化合物进行了测定。f检验(p=0.95,n=3)比较两种方法得到的结果,没有观察到显着差异,检验结果表明超声辅助功能化离体分散液液微萃取的方法用来测定此类样品。
[0055]
7)加标样品测定:为了评估所提出方法的准确性,将三个水平的四种蒽醌类化合物添加到样品溶液中。如表2所示,四种蒽醌类化合物的回收率范围为91.88%至105.27%,精度为2.44
‑
5.67%(rsd)。因此,所有结果表明该方法是一种精确可靠的方法,可以代替常规方法检测实际样品中的这些蒽醌类化合物。
[0056]
表2对于不同浓度加标样品溶液,方法的准确性(相对回收率及其rsd)
[0057][0058][0059]
显然,上述实施例仅仅是为清楚地说明所作的举例,并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1