呋塞米中有关物质的检测方法及应用与流程
1.本发明涉及药物成分分析检测技术领域,具体涉及一种呋塞米中有关物质的检测方法及应用。
背景技术:
2.呋塞米,又名呋喃苯胺酸、速尿,是一种广泛应用于治疗充血性心力衰竭和水肿的袢利尿药。其化学名称为2
‑
[(2
‑
呋喃甲基)氨基]
‑5‑
(氨磺酰基)
‑4‑
氯苯甲酸,分子式为c
12
h
11
cln2o5s,其化学结构式如下式(1)所示:
[0003][0004]
由于呋塞米对光不稳定,尤其在酸性的条件下对光极其敏感,会降解生产诸多有关物质,例如杂质a、杂质b、杂质c、杂质d、杂质e和杂质f。
[0005]
其中,杂质b的化学名称为2,4
‑
二氯
‑5‑
(磺酰胺基)苯甲酸,分子式为c7h5cl2no4s,分子量为270.09,其结构式如式(2)所示:
[0006][0007]
杂质e的化学名称为2,4
‑
二氯苯甲酸,分子式为c7h4cl2o2,分子量为191.01,其结构式如式(3)所示:
[0008][0009]
杂质f的化学名称为4
‑
氯
‑5‑
氨磺酰基
‑2‑
{[(2rs)
‑
四氢呋喃
‑2‑
甲基]氨基}
‑
苯甲酸,分子式为c
12
h
15
cln2o5s,分子量为334.78,其结构式如式(4)所示:
[0010]
[0011]
现有技术中对杂质a、杂质c和杂质d有成熟的方法进行检测控制,而对于呋塞米中的杂质b、杂质e和杂质f检测的研究则相对较少,检测方法目前只局限于高效液相色谱检测,流动相的组分繁多,配制复杂,杂质和目标物质分离效果不理想,而且不能同时检测杂质b、杂质e和杂质f。
[0012]
有鉴于此,特提出本发明。
技术实现要素:
[0013]
本发明的目的之一在于提供一种呋塞米中有关物质的检测方法,解决了现有高效液相色谱检测中流动相的组分繁多,配制复杂,杂质和目标物质分离效果不理想,而且不能同时检测杂质b、杂质e和杂质f的技术问题。
[0014]
本发明的目的之二在于提供一种呋塞米中有关物质的检测方法在呋塞米的原料或制剂的质量控制中的应用,为评价或控制呋塞米的原料或制剂的质量提供依据。
[0015]
为解决上述技术问题,本发明特采用如下技术方案:
[0016]
本发明的第一方面提供了一种呋塞米中有关物质的检测方法,采用高效液相色谱对待测样品溶液进行检测;
[0017]
其中,所述高效液相色谱以流动相a和流动相b的混合溶液作为流动相,进行梯度洗脱。
[0018]
所述流动相a与所述流动相b起始的体积比为88
‑
92:12
‑
8。
[0019]
所述梯度洗脱中,所述流动相a与所述流动相b的体积比从88
‑
92:12
‑
8到12
‑
8:88
‑
92最终回到88
‑
92:12
‑
8。
[0020]
可选地,所述流动相a与所述流动相b起始的体积比为90:10。
[0021]
优选地,所述待测样品溶液中呋塞米浓度为1.0mg/ml。
[0022]
可选地,流动相a包括质量百分比为0.1%的磷酸水溶液;流动相b包括乙腈。
[0023]
可选地,所述流动相a与所述流动相b的体积比从90:10到10:90。
[0024]
优选地,所述梯度洗脱的过程为:
[0025]0‑
20min内,流动相a和流动相b的体积比从90:10到65:35。
[0026]
20
‑
35min内,流动相a和流动相b的体积比从65:35到55:45。
[0027]
35
‑
40min内,流动相a和流动相b的体积比从55:45到10:90。
[0028]
40
‑
45min内,流动相a和流动相b的体积比保持10:90。
[0029]
45
‑
45.1min内,流动相a和流动相b的体积比从10:90到90:10。
[0030]
45.1
‑
55min内,流动相a和流动相b的体积比保持90:10。
[0031]
可选地,所述碳八色谱柱包括辛烷基硅烷键合硅胶。
[0032]
优选地,所述流动相的流速为0.8
‑
1.2ml/min。
[0033]
优选地,所述流动相的流速为1.0ml/min。
[0034]
优选地,所述色谱柱的柱温为30
‑
40℃。
[0035]
优选地,所述色谱柱的柱温为35℃。
[0036]
可选地,进样量为5
‑
40μl。
[0037]
优选地,所述进样量为20μl。
[0038]
优选地,检测波长为220
‑
280nm。
[0039]
优选地,所述检测波长为238nm。
[0040]
可选地,所述待测样品溶液采用稀释剂溶解待测样品。
[0041]
优选地,所述稀释剂为水、乙腈和冰醋酸的混合溶液。
[0042]
优选地,所述稀释剂中水、乙腈和冰醋酸的体积比为500:500:22。
[0043]
可选地,所述待测样品包括含呋塞米的原料或制剂。
[0044]
可选地,通过外标法对所述待测样品中的有关物质的含量进行计算。
[0045]
本发明的第二方面提供了第一方面所述的检测方法在呋塞米的原料或制剂的质量控制中的应用。
[0046]
本发明提供的呋塞米中有关物质的检测方法,优化了流动相组分的配比,适用性好,减少了传统高效液相色谱流动相的多种组分和繁琐的工艺。使用梯度洗脱缩短了分析周期,提高了有关物质的分离能力,峰型得到了改善,增加了灵敏度。该检测方法简捷,能准确地分析测定呋塞米的原料或制剂中的有关物质的含量,实现了对呋塞米中有关物质更为全面的控制,从而保证了呋塞米的质量。
[0047]
本发明提供的呋塞米中有关物质的检测方法在在呋塞米的原料或制剂的质量控制中的应用,为评价或控制呋塞米的原料或制剂的质量提供依据,同时为呋塞米的原料或制剂提供了更为全面的控制,满足了药品注册审评中对于呋塞米的原料或制剂中杂质的安全性评价的需求,也保证了呋塞米的原料或制剂质量可控,有利于长期稳定性存放和质量监控。
附图说明
[0048]
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0049]
图1为实施例1使用的检测条件得到的呋塞米中杂质的高效液相色谱图;
[0050]
图2为实施例2使用的检测条件得到的呋塞米中杂质的高效液相色谱图;
[0051]
图3为实施例3使用的检测条件得到的呋塞米中杂质的高效液相色谱图。
具体实施方式
[0052]
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明实施例的组件可以以各种不同的配制来布置和设计。
[0053]
呋塞米,又名呋喃苯胺酸、速尿,是一种广泛应用于治疗充血性心力衰竭和水肿的袢利尿药。在机体内,呋塞米对水和电解质排泄有影响作用:能增加水、钠、氯、钾、钙、镁、磷等的排泄。与噻嗪类利尿药不同,呋塞米等袢利尿药存在明显的剂量
‑
效应关系。随着剂量加大,利尿效果明显增强,且药物剂量范围较大。本类药物主要通过抑制肾小管髓袢厚壁段对nacl的主动重吸收,结果管腔液na
+
、cl
‑
浓度升高,而髓质间液na
+
、cl
‑
浓度降低,使渗透压梯度差降低,肾小管浓缩功能下降,从而导致水、na
+
、cl
-
排泄增多。由于na
+
重吸收减少,远端小管na
+
浓度升高,促进na
+
‑
k
+
和na
+
‑
h
+
交换增加,k
+
和h
+
排出增多。至于呋塞米抑制肾
小管髓袢升支厚壁段重吸收cl
-
的机制,过去曾认为该部位存在氯泵,目前研究表明该部位基底膜外侧存在与na
+
‑
k
+
atp酶有关的na
+
、cl
-
配对转运系统,呋塞米通过抑制该系统功能而减少na
+
、c1
-
的重吸收。另外,呋塞米可能尚能抑制近端小管和远端小管对na
+
、cl
-
的重吸收,促进远端小管分泌k
+
。呋塞米通过抑制亨氏袢对ca
2+
、mg
2+
的重吸收而增加ca
2+
、mg
2+
排泄。短期用药能增加尿酸排泄,而长期用药则可引起高尿酸血症。
[0054]
呋塞米对血流动力学的影响:呋塞米能抑制前列腺素分解酶的活性,使前列腺素e2含量升高,从而具有扩张血管作用。扩张肾血管,降低肾血管阻力,使肾血流量尤其是肾皮质深部血流量增加,在呋塞米的利尿作用中具有重要意义,也是其用于预防急性肾功能衰竭的理论基础。另外,与其他利尿药不同,袢利尿药在肾小管液流量增加的同时肾小球滤过率不下降,可能与流经致密斑的氯减少,从而减弱或阻断了球
‑
管平衡有关。呋塞米能扩张肺部容量静脉,降低肺毛细血管通透性,加上其利尿作用,使回心血量减少,左心室舒张末期压力降低,有助于急性左心衰竭的治疗。由于呋塞米可降低肺毛细血管通透性,为其治疗成人呼吸窘迫综合征提供了理论依据。
[0055]
呋塞米,在片剂中的规格为20mg每片;在临床上,用于治疗水肿性疾病,高血压,预防急性肾功能衰竭,高钾血症及高钙血症,稀释性低钠血症,抗利尿激素分泌过多症,急性药物毒物中毒等。
[0056]
由于呋塞米对光不稳定,尤其在酸性的条件下对光极其敏感,会降解生产诸多杂质,例如杂质a、杂质b、杂质c、杂质d、杂质e和杂质f。
[0057]
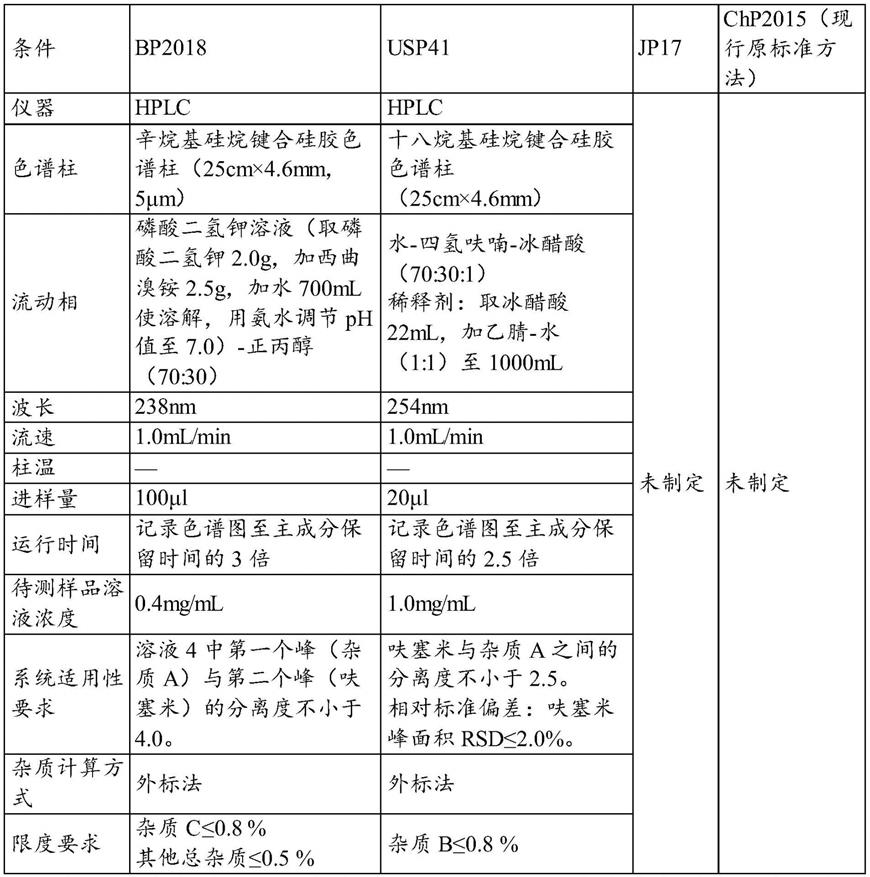
查询国内外药典,对于呋塞米中的杂质检测的研究则相对较少,检测方法目前只局限于高效液相色谱检测,且流动相的组分繁多,配制复杂,杂质和目标物质分离效果不理想。随者国家对人民用药安全有效性的高度重视,在药物注册审批时对于呋塞米中的杂质研究要求日益提高。下表对各国药典中呋塞米片的杂质检测方法及目标杂质的控制情况进行了对比。
[0058]
表1现行版国内外药典杂质检测方法收载情况
[0059][0060]
各国药典杂质检测方法对比可知,usp41呋塞米片杂质采用外标法测定杂质b(限度为≤0.8%),bp2018呋塞米片杂质采用外标法测定杂质c(限度为≤0.8%)和其它总杂质(限度为≤0.5%)。通过杂质结构对比,usp杂质b与bp杂质c为同一杂质。另根据bp2018呋塞米原料药标准可知,呋塞米可能存在的杂质有杂质a、杂质b、杂质c、杂质d、杂质e和杂质f,共计6个。
[0061]
在现有的技术里,对呋塞米的原料或制剂中有关物质的检测方法不全面,有关物质中杂质a、杂质c和杂质d已有成熟的方法进行检测控制,而对于另外的杂质b、杂质e和杂质f则未见成熟方法进行检测控制,不能满足现行药品注册审评中对于杂质的安全性评的需求。
[0062]
根据本发明的第一方面提供的一种呋塞米中有关物质的检测方法,采用高效液相色谱对待测样品溶液进行检测。
[0063]
其中,所述高效液相色谱以流动相a和流动相b的混合溶液作为流动相,进行梯度洗脱。
[0064]
所述流动相a与所述流动相b起始的体积比为88
‑
92:12
‑
8。
[0065]
所述梯度洗脱中,所述流动相a与所述流动相b的体积比从88
‑
92:12
‑
8到12
‑
8:88
‑
92最终回到88
‑
92:12
‑
8。
[0066]
本发明提供的呋塞米中有关物质的检测方法,优化了流动相组分的配比,适用性好,减少了传统高效液相色谱流动相的多种组分和繁琐的工艺。使用梯度洗脱缩短了分析周期,提高了有关物质的分离能力,峰型得到了改善,增加了灵敏度。该检测方法简捷,能准确地分析测定呋塞米的原料或制剂中的有关物质的含量,实现了对呋塞米中有关物质更为全面的控制,从而保证了呋塞米的质量。
[0067]
梯度洗脱是指在同一个分析周期中,按一定程度不断改变流动相a和流动相b的体积比进行洗脱的过程。在液相色谱中对呋塞米样品采用梯度洗脱的方法,从而可以使呋塞米样品中的性质差异较大的组分能按各自适宜的容量因子k达到良好的分离目的。
[0068]
在本发明的一些实施方式中,所述流动相a与所述流动相b起始的体积比典型但非限制性的为88:12、89:11、90:10、91:9或92:8。
[0069]
在本发明的一些实施方式中,梯度洗脱中流动相a与所述流动相b从起始的体积比88
‑
92:12
‑
8到12
‑
8:88
‑
92最终回到88
‑
92:12
‑
8,不断改变流动相a和流动相b体积比,使待测样品中各组分达到分离的目的。
[0070]
在本发明的一种优选实施方式中,所述流动相a与所述流动相b起始的体积比为90:10。
[0071]
优选地,所述待测样品溶液中呋塞米浓度为1.0mg/ml。
[0072]
可选地,流动相a包括质量百分比为0.1%的磷酸水溶液;流动相b包括乙腈。
[0073]
可选地,所述梯度洗脱的过程包括:
[0074]0‑
20min内,流动相a和流动相b的体积比从90:10到65:35;
[0075]
20
‑
35min内,流动相a和流动相b的体积比从65:35到55:45;
[0076]
35
‑
40min内,流动相a和流动相b的体积比从55:45到10:90;
[0077]
40
‑
45min内,流动相a和流动相b的体积比保持10:90;
[0078]
45
‑
45.1min内,流动相a和流动相b的体积比从10:90到90:10;
[0079]
45.1
‑
55min内,流动相a和流动相b的体积比保持90:10。
[0080]
可选地,所述碳八色谱柱包括辛烷基硅烷键合硅胶。
[0081]
可选地,所述流动相的流速为0.8
‑
1.2ml/min。
[0082]
在固定相为辛烷基硅烷键合硅胶和流动相为流动相a和流动相b的混合溶液的条件下,降低流动相的流速可以提高柱效,但是检测时间会变长。在本发明的一些实施方式中,在满足分离效率的前提下,可以适当提高流速在0.8
‑
1.2ml/min。
[0083]
在本发明的一些实施方式中,所述流动相的流速典型但非限制性的为0.8ml/min、0.9ml/min、1.0ml/min、1.1ml/min或1.2ml/min。
[0084]
在本发明的一种优选实施方式中,所述流动相的流速为1.0ml/min。
[0085]
提高色谱柱的温度,可以降低流动相粘度,提高组分传质速度,提高分析速度,但同时会降低分离度,在本发明的实施方式中,色谱柱的柱温为30
‑
40℃。
[0086]
在本发明的一些实施方式中,所述色谱柱的柱温典型但非限制性的为30℃、31℃、32℃、33℃、34℃、35℃、36℃、37℃、38℃、39℃或40℃。
[0087]
在本发明的一种优选实施方式中,所述色谱柱的柱温为35℃。
[0088]
进样量大可以提高检测灵敏度和准确度,但会降低分离度,同时会更容易污染色
谱柱。在本发明的实施方式中,进样量为5
‑
40μl。
[0089]
在本发明的一些实施方式中,进样量典型但非限制性的为5μl、10μl、15μl、20μl、25μl、30μl、35μl或40μl。
[0090]
在本发明的一种优选实施方式中进样量为20μl。
[0091]
优选地,检测波长为220
‑
280nm。
[0092]
在波长为220
‑
280nm,呋塞米中杂质b、杂质e和杂质f均能达到较好的吸收,同时杂质b、杂质e和杂质f的检测限和灵敏度都能满足要求。
[0093]
在本发明的一些实施方式中,检测波长典型但非限制性的为220nm、230nm、238nm、240nm、250nm、260nm、270nm或280nm,优选的检测波长为238nm。
[0094]
可选地,所述待测样品溶液采用稀释剂溶解待测样品。
[0095]
优选地,所述稀释剂为水、乙腈和冰醋酸的混合溶液。
[0096]
优选地,所述稀释剂中水、乙腈和冰醋酸的体积比为500:500:22。
[0097]
可选地,所述待测样品包括含呋塞米的原料或制剂。
[0098]
在本发明的一些实施方式中,含呋塞米的原料或制剂典型但非限制性的为呋塞米注射液、呋塞米片、呋塞米颗粒、呋塞米缓释胶囊、呋塞米口服液。
[0099]
可选地,通过外标法对所述待测样品中的有关物质的含量进行计算。
[0100]
所述外标法是指:按各品种项下的规定,精密称取对照品和待测样品,配制成溶液,分别精密取一定量,进样,记录色谱图,测量对照品溶液和待测样品溶液中待测物质的峰面积,按下式(5)计算:
[0101]
含量(c
x
)=a
x
*c
r
/a
r
[0102]
式(5)
[0103]
式(5)中:a
x
为待测样品的峰面积;a
r
为对照品的峰面积;c
x
为待测样品溶液的浓度;c
r
为对照品的浓度;可得到相应呋塞米片中杂质b、杂质e、杂质f的含量。
[0104]
根据本发明的第二方面提供的第一方面所述的检测方法在呋塞米的原料或制剂的质量控制中的应用。
[0105]
本发明提供的呋塞米中有关物质的检测方法在在呋塞米的原料或制剂的质量控制中的应用,为评价或控制呋塞米的原料或制剂的质量提供依据,同时为呋塞米的原料或制剂提供了更为全面的控制,满足了药品注册审评中对于呋塞米的原料或制剂中杂质的安全性评价的需求,也保证了呋塞米的原料或制剂质量可控,有利于长期稳定性存放和质量监控。
[0106]
下面结合实施例和对比例对本发明做进一步详细的说明。
[0107]
实施例使用的试剂和仪器如下:
[0108]
试剂与试药:杂质b对照品、杂质e对照品、杂质f对照品、磷酸、乙腈、冰醋酸。
[0109]
仪器与器具:电子分析天平(十万分之一)、量筒、容量瓶、超声溶解仪器(昆山超声仪器公司kq
‑
500b)、高效液相色谱仪(agilent1260)、色谱柱:agilent eclipse plus c8(250mm
×
4.6mm,5μm)。
[0110]
实施例1
[0111]
1、呋塞米有关物质的检测
[0112]
1.1配制稀释剂:取冰醋酸22ml,置于1000ml容量瓶中,用乙腈与水体积比为1:1的
乙腈水溶液稀释至刻度,混合均匀。
[0113]
1.2对照品溶液配制:分别精密称定10mg的杂质b、杂质e和杂质f的对照品,分别用100ml稀释剂溶解并制成杂质b、杂质e和杂质f的浓度均为0.1mg/ml的溶液。精密量取各溶液1ml,分别置于100ml容量瓶中,用稀释剂稀释至刻度,摇匀,分别得到杂质b的对照品溶液、杂质e的对照品溶液和杂质f的对照品溶液。
[0114]
1.3待测样品溶液配制:在避光条件下将呋塞米片制成粉末,按照呋塞米含量为10mg换算呋塞米片质量进行称取,置于10ml容量瓶中,先加入稀释剂5ml,使用超声溶解仪超声15分钟使呋塞米粉末溶解,放冷后用稀释剂稀释至刻度,混合均匀后使用0.45μm的滤膜过滤,得到的续滤液为待测样品溶液。
[0115]
1.4测定:将配制的对照品溶液和待测样品溶液注入高效液相色谱仪,高效液相色谱仪使用的色谱柱为辛烷基硅烷键合硅胶(250mm
×
4.6mm,5μm);
[0116]
检测波长为238nm;流动相流速为1.0ml/min;
[0117]
柱温:35℃;进样量:20μl;
[0118]
流动相为流动相a和流动相b的混合溶液;
[0119]
流动相a为质量百分比为0.1%的磷酸水溶液;
[0120]
流动相b为乙腈;
[0121]
所述流动相a和流动相b的体积比例按照下表2进行梯度洗脱;
[0122]
表2梯度洗脱数据时间表
[0123]
时间(分钟)流动相a(%)流动相b(%)0901020653535554540109045109045.19010559010
[0124]
2、呋塞米有关物质检测方法验证
[0125]
2.1标准曲线
[0126]
2.1.1制作标准曲线:精密称取杂质b、杂质e和杂质f分别配制成0.01μg/ml、0.1μg/ml、0.5μg/ml、0.8μg/ml、1.0μg/ml、2.0μg/ml和5.0μg/ml的标准溶液,分别取各浓度溶液20μl,注入高效液相色谱仪,记录色谱图,以浓度对峰面积进行线性回归,计算回归方程、相关系数及截距,得到的杂质b、杂质e和杂质f的标准曲线数据如表4
‑
6所示。
[0127]
表4杂质b的标准曲线数据
[0128]
[0129]
结论:杂质b浓度在0.0141μg/ml
‑
5.0444μg/ml(相当于待测样品溶液浓度的0.001%~0.5%)范围内呈良好线性关系,相关系数为0.9998,截距为2.16%,可以用于测定杂质b的含量。
[0130]
表5杂质e的标准曲线数据
[0131][0132]
结论:杂质e浓度在0.0137μg/ml
‑
5.0234μg/ml(相当于待测样品溶液浓度的0.001%~0.5%)范围内呈良好线性关系,相关系数为0.9998,截距为0.98%,可以用于测定杂质e的含量。
[0133]
表6杂质f的标准曲线数据
[0134][0135]
结论:杂质f浓度在0.0056μg/ml
‑
5.1992μg/ml(相当于待测样品溶液浓度的0.0006%~0.5%)范围内呈良好线性关系,相关系数为0.9998,截距为2.32%,可以用于测定杂质f的含量。
[0136]
2.1.2测定结果
[0137]
将配制的待测样品溶液注入高效液相色谱仪,得到待测样品溶液的液相色谱图,与对照品溶液的液相色谱图进行比较,根据相对保留时间定性,色谱峰面积定量,图1为本实施例得到的待测样品溶液的液相色谱图。通过标准曲线计算得出:杂质b的质量浓度为0.1mg/ml;杂质c的质量浓度为0.1mg/ml;杂质f的质量浓度为0.1mg/ml。
[0138]
2.2检测限与定量限的检测
[0139]
2.2.1根据系统精密度项下的等量混合对照品溶液色谱图,分别计算各杂质峰的s/n值,按照信噪比3:1确定检测限,按照信噪比10:1确定定量限。结果见表7所示。
[0140]
表7定量限及检测限的试验结果
[0141][0142]
结论:各杂质的定量限均小于1
×
10
‑7g/ml(相当于供试品溶液浓度的0.01%),说明本方法的灵敏度高。
[0143]
2.2.2检测限与定量限的确认
[0144]
根据表7测量的杂质b、杂质e和杂质f得到的定量限浓度,配制成相应浓度的标准
溶液,按照与1.4中同样的色谱系统进样分析,同时采集基线噪声,按照信噪比10
±
1确认定量限,信噪比3
±
1确认检测限。结果见表8所示。
[0145]
表8检测限与定量限确认试验结果
[0146][0147]
结论:各杂质定量限浓度及检测限浓度确认的信噪比符合要求。
[0148]
2.2.3定量限的精密度
[0149]
根据表7测量的杂质b、杂质e和杂质f得到的定量限浓度,配制成相应浓度的标准溶液,按照与1.4中同样的色谱系统进样分析,连续进样6针,记录色谱图。计算各峰面积的rsd。结果见表9所示:
[0150]
表9定量限精密度试验结果
[0151]
编号/峰面积杂质b杂质f杂质e10.77250.71981.383120.77660.72021.566530.73470.79151.795440.73290.80001.421850.67400.72851.566860.74980.77411.6222平均值0.740.761.56rsd(%)5.034.889.50
[0152]
结论:各杂质峰面积rsd小于10.0%,说明定量限精密度良好。
[0153]
2.2.4定量限的重复性
[0154]
根据表7测量的杂质b、杂质e和杂质f得到的定量限浓度,配制成相应浓度的标准溶液,分为6份,按照与1.4中同样的色谱系统进样分析,记录色谱图,计算各杂质峰的响应因子(浓度与峰面积之比)。结果见表10所示:
[0155]
表10定量限重复性试验结果
[0156]
编号/响应因子杂质b杂质f杂质e11.76
×
10
‑86.68
×
10
‑98.61
×
10
‑921.91
×
10
‑88.83
×
10
‑98.68
×
10
‑931.89
×
10
‑87.71
×
10
‑98.85
×
10
‑941.81
×
10
‑87.58
×
10
‑98.77
×
10
‑951.71
×
10
‑87.44
×
10
‑99.48
×
10
‑961.83
×
10
‑87.94
×
10
‑99.27
×
10
‑9平均值1.82
×
10
‑87.70
×
10
‑98.94
×
10
‑9rsd(%)4.189.103.92
[0157]
结论:各杂质的响应因子rsd均小于10%,符合测定要求。
[0158]
3、回收试验
[0159]
3.1在避光条件下将呋塞米片制成粉末,按照呋塞米含量为10mg换算呋塞米片质量进行称取10份,置于10个100ml容量瓶中。
[0160]
3.2对照品溶液配制:分别精密称定10mg的杂质b、杂质e和杂质f的对照品,分别置于100ml容量瓶中,用稀释剂稀释至刻度,摇匀,分别得到浓度均为100μg/ml的杂质b的对照品贮备溶液、杂质e的对照品贮备溶液和杂质f的对照品贮备溶液。
[0161]
3.3取3.1中的容量瓶3个,分别精密加入杂质b的对照品贮备溶液、杂质e的对照品贮备溶液和杂质f的对照品贮备溶液各0.1ml,加入稀释剂,超声15min,稀释至刻度,摇匀,滤过,取续滤液分别作为待测样品溶液;使各杂质浓度约相当于待测样品溶液浓度的0.01%。
[0162]
3.4取3.1中的容量瓶3个,分别精密加入杂质b的对照品贮备溶液、杂质e的对照品贮备溶液和杂质f的对照品贮备溶液各1.0ml,加入稀释剂,超声15min,稀释至刻度,摇匀,滤过,取续滤液分别作为待测样品溶液;使各杂质浓度约相当于待测样品溶液浓度的0.10%。
[0163]
3.5取3.1中的容量瓶3个,分别精密加入杂质b的对照品贮备溶液、杂质e的对照品贮备溶液和杂质f的对照品贮备溶液各1.5ml,加入稀释剂,超声15min,稀释至刻度,摇匀,滤过,取续滤液分别作为待测样品溶液;使各杂质浓度约相当于待测样品溶液浓度的0.15%。
[0164]
3.6将3.1剩余的1个容量瓶用稀释剂溶解并稀释至刻度,即得空白底样。
[0165]
3.7精密称取杂质b、e、f对照品贮备液各1ml,置100量瓶中,用稀释剂稀释至刻度制成每1ml中含1μg的混合溶液作为对照品溶液。分别精密量取待测样品溶液和对照品溶液20μl进样检测,记录色谱图。按外标法计算每个待测样品溶液中各杂质含量,根据加入量和样品本身的杂质量计算回收率,结果如表11
‑
13所示。
[0166]
表11杂质b回收率验证结果
[0167][0168][0169]
结论:本法测定的杂质b回收率为90.14%
‑
117.46%,平均回收率为102.50%,rsd为8.28%,本方法能够准确测定本品中杂质b的含量。
[0170]
表12杂质e回收率验证结果
[0171][0172]
结论:本法测定的杂质e回收率为107.14%
‑
116.36%,平均回收率为111.66%,rsd为3.96%,本方法能够准确测定本品中杂质e的含量。
[0173]
表13杂质f回收率验证结果
[0174][0175]
结论:本法测定的杂质f回收率为97.75%
‑
119.90%,平均回收率为105.81%,rsd为5.91%,本方法能够准确测定本品中杂质f的含量。
[0176]
4、溶液稳定性
[0177]
4.1待测样品溶液:在避光条件下将呋塞米片制成粉末,按照呋塞米含量为10mg换算呋塞米片质量进行称取,置于10ml容量瓶中,先加入稀释剂5ml,使用超声溶解仪超声15分钟使呋塞米粉末溶解,放冷后用稀释剂稀释至刻度,混合均匀后使用0.45μm的滤膜过滤,得到的续滤液为待测样品溶液。
[0178]
4.2对照品溶液:分别精密称定1mg的杂质b、杂质e和杂质f的对照品,分别用10ml稀释剂溶解并制成杂质b、杂质e和杂质f的浓度均为0.1mg/ml的溶液。精密量取各溶液1ml,分别置于100ml容量瓶中,用稀释剂稀释至刻度,摇匀,分别得到杂质b的对照品溶液、杂质e的对照品溶液和杂质f的对照品溶液。
[0179]
4.3精密量4.2得到的对照品溶液、杂质e的对照品溶液和杂质f的对照品溶液,分别在0h、2h、4h、8h、12h、24h后按照1.4所述的色谱条件进样,记录色谱图,得到的结果如表14所示。
[0180]
表14各杂质对照品溶液稳定性试验数据表
[0181]
放置时间(h)/峰面积杂质b杂质e杂质f0h51.875460.8825122.4061
2h52.049460.9248122.66594h52.229261.1196122.92898h52.519261.3679123.721512h52.779461.7143124.461724h54.795664.0945131.7314峰面积均值52.7161.68124.65rsd(%)2.041.982.85
[0182]
结论:对照品溶液中各杂质对照品的峰面积rsd小于5.0%,说明各杂质在24小时内稳定。
[0183]
取4.1得到的待测样品溶液分别在0h、2h、4h、8h、12h、24h后按照1.4部分所述的色谱条件进样,记录色谱图,得到的结果如表15所示。
[0184]
表15待测样品溶液稳定性试验数据表
[0185][0186][0187]
结论:由上述数据可以看出,杂质f在24小时检出量一致,杂质b、e均未检出,本品稳定性较好。
[0188]
5、重复性
[0189]
5.1待测样品溶液:在避光条件下将呋塞米片制成粉末,按照呋塞米含量为10mg换算呋塞米片质量进行称取,置于10ml容量瓶中,先加入稀释剂5ml,使用超声溶解仪超声15分钟使呋塞米粉末溶解,放冷后用稀释剂稀释至刻度,混合均匀后使用0.45μm的滤膜过滤,得到的续滤液为待测样品溶液。
[0190]
5.2对照品溶液:分别精密称定1mg的杂质b、杂质e和杂质f的对照品,分别用10ml稀释剂溶解并制成杂质b、杂质e和杂质f的浓度均为0.1mg/ml的溶液。精密量取各溶液1ml,分别置于100ml容量瓶中,用稀释剂稀释至刻度,摇匀,分别得到杂质b的对照品溶液、杂质e的对照品溶液和杂质f的对照品溶液。
[0191]
5.3取5.1得到的待测样品溶液和5.2得到的对照品溶液各6份,每份20μl,分别按照1.4部分所述的色谱条件进样,记录色谱图,得到的结果如表16所示。
[0192]
表16重复性试验数据表
[0193][0194][0195]
结论:平行测定待测样品溶液6份,杂质b、e均未检出,杂质f测定结果一致,表明杂质测定方法重复性良好。
[0196]
6、中间精密度
[0197]
本试验例对不同分析人员和仪器对测试结果的精密度进行考察,得到中间精密度数据。取5.1得到的待测样品溶液12份,每份20μl,分成2组,由两名分析人员分别按照1.4部分所述的色谱条件选用不同的高效液相色谱仪进样,1#分析人员使用的高效液相色谱仪的型号为agilent1260,厂家为安捷伦公司;2#分析人员使用的高效液相色谱仪的型号为agilent1260,厂家为安捷伦公司,记录得到的色谱图,得到的中间精密度数据如表17所示。
[0198]
表17中间精密度数据表
[0199][0200]
实施例2
[0201]
本实施例对呋塞米有关物质进行检测,按照实施例1中的呋塞米有关物质的检测步骤进行,与实施例1不同的是:1.4测定中流动相流速为1.2ml/min,其余步骤和工艺条件均与实施例1的呋塞米有关物质检测部分相同,在此不再赘述,图2为本实施例得到的待测样品溶液的液相色谱图。
[0202]
实施例3
[0203]
本实施例对呋塞米有关物质进行检测,按照实施例1中的呋塞米有关物质的检测步骤进行,与实施例1不同的是:1.4测定中流动相流速为0.8ml/min,其余步骤和工艺条件均与实施例1的呋塞米有关物质检测部分相同,在此不再赘述,图3为本实施例得到的待测样品溶液的液相色谱图。
[0204]
综上所述,本发明提供的一种呋塞米中有关物质的检测方法,能够将呋塞米峰和杂质b、e、f的峰达到有效分离,准确地测定呋塞米中杂质b、杂质e和杂质f的含量,且流速耐用性好,实现了对呋塞米中杂质b、杂质e和杂质f含量的控制,从而保证了呋塞米原料或制剂的质量。本发明填补了现有技术中的空白,有效克服了现有技术中的种种缺点而且具有高度产业利用价值。
[0205]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1