一种测定羟丙纤维素中羟丙氧基含量的方法与流程
1.本发明涉及一种氟离子化合物,尤其涉及一种氟离子化合物的检测方法。
背景技术:
2.羟丙纤维素是一种多用途的非离子型纤维素衍生物,是由碱性纤维素与环氧丙烷在高温高压下反应而得到的非离子型纤维素醚,根据其取代基羟丙氧基含量的不同,分为低取代羟丙纤维素和高取代羟丙纤维素。低取代羟丙纤维素(l-hpc)是白色或类白色纤维状颗粒状粉末,无味无臭或微有异臭且无味。平均粒子的大小不一,其相对粉体性质也不相同,大粒子的崩解性能好,而小粒子的粘结性能突出。l-hpc在水和有机溶剂中不易溶解,但由于其粉体有很大的表面积和孔隙率,故而增大了其吸湿性和吸水量,这种性质大大增加了它的膨胀度,可用作片剂中很好的崩解剂,提高片剂的生物利用度。另外,l-hpc粗糙结构使其能与药物和颗粒之间产生较大的镶嵌作用,增加黏结度,提高成型性,提高片剂硬度和光泽度及外观质量。综上,羟丙纤维素作为崩解剂和粘合剂的特点是容易压制成型,适应性强,特别是对于不易成型、易松片、塑形和脆性大的固体制剂,加入l-hpc就能提高片剂的硬度和光泽度,还能使片剂快速崩解。该产品与多数药物不起任何反应,是一种中性的,对人体无害的纤维素衍生物。
3.因为最终要施用到人体上,因此本领域对控制药物原料的质量控制有着非常严苛的要求,其中就包括控制药用辅料的质量。如前所述,羟丙纤维素作为常用的药用辅料,检测其中羟丙氧基的含量对控制其质量有着重要的意义。
4.现有技术中,用于检测羟丙纤维素中羟丙氧基的方法主要为容量法和气相色谱法。容量法操作简便,但灵敏度不高。中国药典2020版四步披露了一种采用气相色谱法检测羟丙纤维素中羟丙氧基的方法,但样品的处理较为繁琐,且在上样之前需要分离上清液,操作不当容易造成污染。中国非专利文献“顶空气相色谱-质谱法测定低取代羟丙纤维素中羟丙氧基的含量”(张喜金,等,食品安全质量检测学报,2017,第8卷第10期:4065-4068)也披露了一种测试方法,选择顶空进样,虽然部分解决了样品处理的问题,但采用质谱作为检测器,对仪器要求高,不便于推广使用。
5.因此,如何快速、简便、精确地测定羟丙纤维素中羟丙氧基的含量是一个亟待解决的技术难题。
技术实现要素:
6.本发明所要解决的技术问题是提供一种测定羟丙纤维素中羟丙氧基含量的方法,该方法操作简便、专属性强、灵敏度高、重复性好。
7.为了解决上述技术问题,本发明采用如下技术方案:
8.一种测定羟丙纤维素中羟丙氧基含量的方法,所述的方法为气相色谱法。
9.进一步的,所述方法的条件如下:
10.氢火焰离子化检测器;0.53mm
×
30m玻璃毛细管柱,用6%氰丙基苯基-94%二甲基
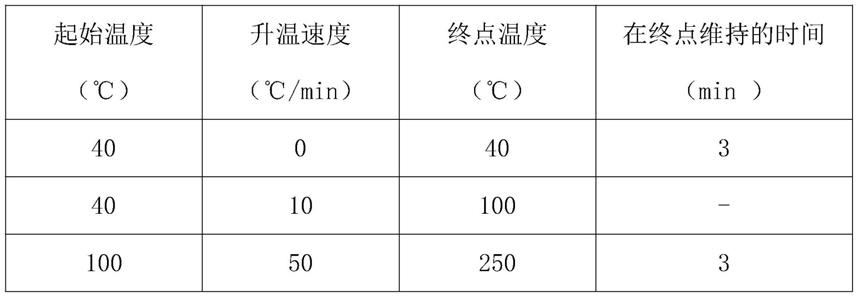
硅氧烷为固定液,固定液的厚度为3μm。
11.进一步的,所述方法的色谱条件如下:
12.顶空进样器,进样量2μl,载气为氦,流速1.5ml/min,分流比50:1;
13.进样器温度180℃,检测器温度280℃;
14.色谱时长:15min;
15.色谱柱程序升温条件:
16.。
17.进一步的,所述方法的系统适用性如下:
18.理论塔板数不低于10000;
19.对照品峰与内标物质峰应完全分离;
20.取对照品溶液2μl注入气相色谱仪,连续进样6次,计算校正因子,相对标准偏差应不大于3.0%。
21.进一步的,所述的方法包括如下步骤:
22.内标溶液:取1ml甲基环己烷溶于50ml邻二甲苯中,即得;
23.标准品溶液:称取60mg己二酸于顶空瓶中,加入内标溶液2ml,57%hi溶液1.0ml,密封,准确称重;然后利用微量注射器穿刺加入25μl2-碘丙烷,精密称定,计算前后2次称重差值得到加入2-碘丙烷的质量;
24.供试品溶液:精确称取30mg羟丙纤维素于顶空瓶中,加入20mg己二酸,内标溶液2ml,57%hi溶液1.0ml,密封;115
±
2℃反应70min,每隔20min,振摇1min,冷却至室温,用注射器加入1.0m l 8mol/l naoh溶液,充分振荡,中和剩余的hi。
25.进一步的,计算羟丙氧基含量的方法如下:
26.(1)计算响应因子f
27.f=(a1
×w×
c)/(a2
×
100)
28.其中:a1=标准品溶液中内标物的峰面积;
29.w=准品溶液中2-碘丙烷的质量,mg;
30.c=2-碘丙烷的含量,%;
31.a2=标准溶液中2-碘丙烷的峰面积;
32.(2)计算羟丙氧基的百分比含量,mg/mg,%;
33.result=(a4
×f×
m1
×
1.15
×
100)/(a3
×
w2
×
m2)
34.其中:a4=供试品溶液中2-碘丙烷的峰面积;
35.f=响应因子;
36.m1=羟丙氧基分子量,75.1;
37.1.15=校正因子;
38.a3=供试品溶液中内标物的峰面积;
39.w2=供试品溶液中供试品的质量,mg;
40.m2=2-碘丙烷的分子量,166。与现有技术相比,本发明的有益效果主要体现在如下几个方面:
41.1.本发明方法操作简便。
42.2.本发明方法专属性强。
43.3.本发明方法重复性好。
44.4.本发明方法准确度高。
45.5.本发明方法精密度高。
46.本发明方法的技术效果可以通过以下试验进行证明。
47.试验例 方法学验证
48.1.专属性
49.反应瓶中加入己二酸20mg,分别精密加入邻二甲苯2.0ml、氢碘酸各1.0ml和8mol/lnaoh溶液1.0ml,密封,115
±
2℃反应70min,通过顶空进样。结果显示,在本发明选定的色谱条件下,空白溶液无干扰,说明本发明方法专属性好。
50.2.线性范围
51.取标准品加水制成母液,稀释成系列浓度,依本发明方法测定2-碘甲烷的峰面积,结果见表1,结果表明2-碘甲烷浓度在0.5-16.0mg/ml范围内与峰面积线性关系良好。
52.表1本发明方法线性试验
[0053][0054]
3.加样回收率
[0055]
取同一批羟丙纤维素,加入2-碘甲烷标准品,加入量与供试品中待测成分的含量之比控制在1-2:1左右,注入气相色谱仪,依法测定含量,计算回收率。结果见表2,结果表明本发明方法准确度良好。
[0056]
表2加样回收率
[0057]
[0058][0059]
4.重复性
[0060]
取同一批羟丙纤维素,依本发明方法连续测定其中的羟丙氧基含量,结果见表3,结果表明,本发明方法重复性良好。
[0061]
表3本发明方法重复性
[0062]
次数123456avrsd结果(%)67.566.967.467.667.367.867.40.45
[0063]
5.中间精密度
[0064]
取同一批羟丙纤维素,由两个不同的测试人员在不同的时间测定其中的羟丙氧基含量,结果见表4,结果表明,本发明方法的中间精密度良好。
[0065]
表4本发明方法中间精密度
[0066]
具体实施方式
[0067]
实施例仅用于示例性说明,不能理解为对本专利的限制。
[0068]
实施例羟丙纤维素中羟丙氧基含量的测定
[0069]
采用气相色谱法。
[0070]
色谱条件如下:
[0071]
氢火焰离子化检测器;0.53mm
×
30m玻璃毛细管柱,用6%氰丙基苯基-94%二甲基
硅氧烷为固定液,固定液的厚度为3μm;
[0072]
顶空进样器,进样量2μl,载气为氦,流速1.5ml/min,分流比50:1;
[0073]
进样器温度180℃,检测器温度280℃;
[0074]
色谱时长:15min;
[0075]
色谱柱程序升温条件:
[0076][0077]
系统适用性如下:
[0078]
理论塔板数不低于10000;对照品峰与内标物质峰应完全分离;取对照品溶液2μl注入气相色谱仪,连续进样6次,计算校正因子,相对标准偏差应不大于3.0%。
[0079]
内标溶液:取1ml甲基环己烷溶于50ml邻二甲苯中,即得;
[0080]
标准品溶液:称取60mg己二酸于顶空瓶中,加入内标溶液2ml,57%hi溶液1.0ml,密封,准确称重;然后利用微量注射器穿刺加入25μl2-碘丙烷,精密称定,计算前后2次称重差值得到加入2-碘丙烷的质量;
[0081]
供试品溶液:精确称取30mg羟丙纤维素于顶空瓶中,加入20mg己二酸,内标溶液2ml,57%hi溶液1.0ml,密封;115
±
2℃反应70min,每隔20min,振摇1min,冷却至室温,用注射器加入1.0m l 8mol/l naoh溶液,充分振荡,中和剩余的hi。
[0082]
采用下式计算羟丙氧基含量的方法如下:
[0083]
(1)计算响应因子f
[0084]
f=(a1
×w×
c)/(a2
×
100)
[0085]
其中:a1=标准品溶液中内标物的峰面积;
[0086]
w=准品溶液中2-碘丙烷的质量,mg;
[0087]
c=2-碘丙烷的含量,%;
[0088]
a2=标准溶液中2-碘丙烷的峰面积;
[0089]
(2)计算羟丙氧基的百分比含量,mg/mg,%;
[0090]
result=(a4
×f×
m1
×
1.15
×
100)/(a3
×
w2
×
m2)
[0091]
其中:a4=供试品溶液中2-碘丙烷的峰面积;
[0092]
f=响应因子;
[0093]
m1=羟丙氧基分子量,75.1;
[0094]
1.15=校正因子;
[0095]
a3=供试品溶液中内标物的峰面积;
[0096]
w2=供试品溶液中供试品的质量,mg;
[0097]
m2=2-碘丙烷的分子量,166。
[0098]
显然,上述实施例仅仅是为清楚地说明技术方案而作的举例,并非对本发明实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1