一种乙酰半胱氨酸及其有关物质的检测方法与流程
氨基-3-亚磺基丙酸一水合物、(r)-2-乙酰氨基-3-(((r)-2-氨基-2-羧乙基)二硫烷基)丙酸、n-(2-巯基乙基)乙酰胺。
11.可选地,所述色谱柱为ace comixsil acrp色谱柱。
12.可选地,所述梯度洗脱的条件为:
13.从0分钟到20分钟流动相a的体积占比从95-100%逐渐变化为15-25%,流动相b的体积占比从0-5%逐渐变化为75-85%;
14.从20分钟到22分钟流动相a的体积占比从15-25%逐渐变化为95-100%,流动相b的体积占比从75-85%逐渐变化为0-5%;
15.自22分钟始保持流动相a的体积占比为95-100%,流动相b的体积占比为0-5%;
16.自32分钟始保持流动相a的体积占比为95-100%,流动相b的体积占比为0-5%直至检测结束。
17.可选地,所述梯度洗脱的条件为:
18.从0分钟到20分钟流动相a的体积占比从100%逐渐变化为20%,流动相b的体积占比从0%逐渐变化为80%;
19.从20分钟到22分钟流动相a的体积占比从20%逐渐变化为100%,流动相b的体积占比从80%逐渐变化为0%;
20.自22分钟始保持流动相a的体积占比为100%;
21.自32分钟始保持流动相a的体积占比为100%直至检测结束。
22.可选地,所述流动相a中水与磷酸的体积比为100:0.03,所述流动相b中乙腈与磷酸的体积比为100:0.03。
23.可选地,所述高效液相色谱的流速为0.8-1.2ml/min,柱温为25-35℃,检测波长为220nm,进样温度为5℃。
24.可选地,所述高效液相色谱的柱温为30℃。
25.可选地,所述检测方法具体包括以下步骤:
26.将待检测的乙酰半胱氨酸样品溶解于盐酸中并以稀释剂稀释制成第一浓度的供试品溶液;
27.将乙酰半胱氨酸对照品溶解于所述稀释剂中制成第二浓度的对照品溶液;
28.精密称量乙酰胺胱氨酸和各有关物质适量,并共同溶解于所述稀释剂中制成含第三浓度的乙酰半胱氨酸和第四浓度的各有关物质的系统适用性溶液;
29.取所述系统适用性溶液进样进行高效液相色谱分析,以确定乙酰半胱氨酸和各所述有关物质的保留时间;
30.分别取所述供试品溶液和所述对照品溶液进样进行高效液相色谱分析,通过加校正因子的主成分外标法计算所述供试品溶液中乙酰半胱氨酸和各有关物质的含量。
31.可选地,所述第三浓度与所述第四浓度的比例在150:1至250:1范围内;
32.所述第一浓度在所述第三浓度的
±
10%范围内;
33.所述第二浓度在所述第四浓度的
±
10%范围内。
34.本发明提供的乙酰半胱氨酸及其有关物质的检测方法中,有关物质包括乙酰半胱氨酸生产和储存过程中引入的杂质和/或降解产物,通过高效液相色谱将乙酰半胱氨酸及其有关物质进行分离和检测,其中高效液相色谱采用以阴阳离子嵌入修饰烷基键合硅胶为
固定相的色谱柱,且采用梯度洗脱方式,可实现乙酰半胱氨酸及其杂质和降解产物的完全分离,从而提高了乙酰半胱氨酸及有关物质检测的灵敏度和检测的准确度。本发明的方法专属性强、准确度好、精密度高、重复性好,符合药物质量研究标准的技术要求,所得结果稳定可靠。
35.进一步地,本发明提供的乙酰半胱氨酸及其有关物质的检测方法中,采用的色谱柱的固定相耐水性强,并且,采用的流动相对色谱柱的损耗小,可以延长色谱柱的使用寿命,从而降低检测成本。
36.上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,而可依照说明书的内容予以实施,并且为了让本发明的上述和其它目的、特征和优点能够更明显易懂,以下特举本发明的具体实施方式。
37.根据下文结合附图对本发明具体实施例的详细描述,本领域技术人员将会更加明了本发明的上述以及其他目的、优点和特征。
附图说明
38.通过阅读下文优选实施方式的详细描述,各种其他的优点和益处对于本领域普通技术人员将变得清楚明了。附图仅用于示出优选实施方式的目的,而并不认为是对本发明的限制。而且在整个附图中,用相同的参考符号表示相同的部件。在附图中:
39.图1示出了根据本发明一实施例的乙酰半胱氨酸及其有关物质的检测方法的流程示意图;
40.图2示出了根据本发明实施例1的以系统适用性溶液进行高效液相色谱分析得到的色谱图;
41.图3为对比例1中以系统适用性溶液进行高效液相色谱分析得到的色谱图;
42.图4为对比例2中以系统适用性溶液进行高效液相色谱分析得到的色谱图;
43.图5为对比例3中以系统适用性溶液进行高效液相色谱分析得到的色谱图;
44.图6为对比例4中以系统适用性溶液进行高效液相色谱分析得到的色谱图。
具体实施方式
45.下面将参照附图更详细地描述本公开的示例性实施例。虽然附图中显示了本公开的示例性实施例,然而应当理解,可以以各种形式实现本公开而不应被这里阐述的实施例所限制。相反,提供这些实施例是为了能够更透彻地理解本公开,并且能够将本公开的范围完整的传达给本领域的技术人员。
46.发明人经过深入研究,发现乙酰半胱氨酸原料药在生产过程和储存过程中容易引入l-胱氨酸、l-半胱氨酸、n,n
′‑
二乙酰胱氨酸等多种有关物质。除此之外,发明人还按照药品申报注册要求研究了光照、高温和氧化等影响因素试验和酸碱破坏等强制降解试验,发现了多种降解杂质,如(2r)-2-氨基-3-亚磺基丙酸一水合物、(r)-2-乙酰氨基-3-(((r)-2-氨基-2-羧乙基)二硫烷基)丙酸、n-(2-巯基乙基)乙酰胺等。现有技术不能将乙酰半胱氨酸原料药的有关物质以及降解杂质分离开,导致检测不准确。
47.为解决或至少部分解决上述问题,本发明提出了一种乙酰半胱氨酸及其有关物质的检测方法。本文中提及的有关物质可包括乙酰半胱氨酸生产和储存过程中引入的杂质
(如副产物)和/或降解产物。
48.本发明提出的乙酰半胱氨酸及其有关物质的检测方法通过高效液相色谱(hplc)将乙酰半胱氨酸及其有关物质进行分离和检测。特别地,高效液相色谱采用以阴阳离子嵌入修饰烷基键合硅胶为固定相的色谱柱,并采用磷酸水溶液为流动相a和磷酸乙腈溶液为流动相b进行梯度洗脱,以将乙酰半胱氨酸及其有关物质完全分离。阴阳离子嵌入修饰烷基键合硅胶是一种阴阳离子同时嵌入烷基进行修饰的键合硅胶,能够提供稳定的阴离子与阳离子交换作用(阳离子与阴离子排阻作用),从而提升分离能力。
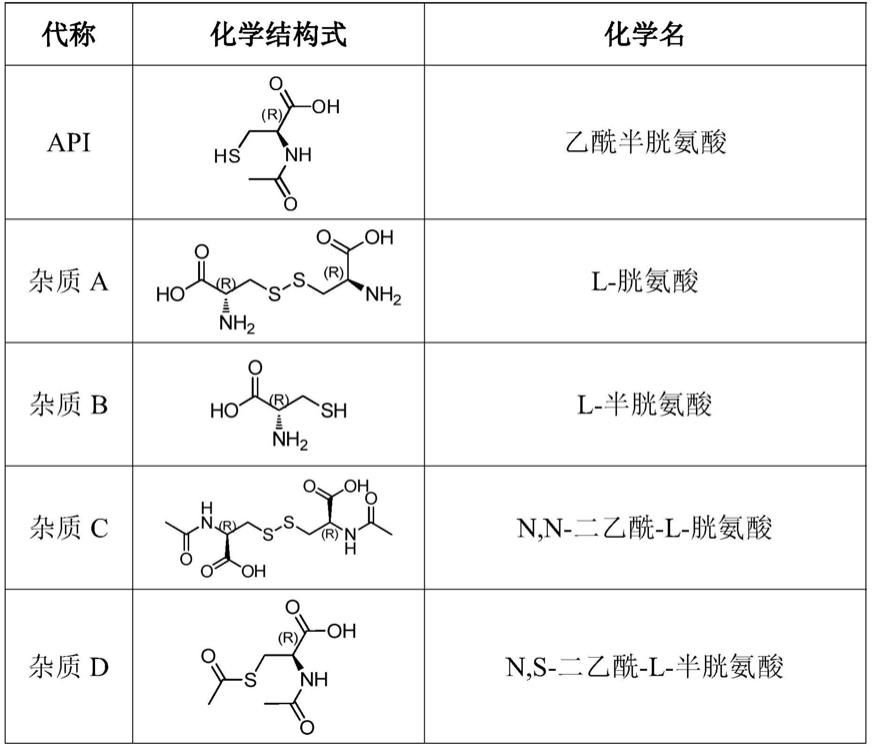
49.本发明实施例提供的乙酰半胱氨酸及其有关物质的检测方法可实现乙酰半胱氨酸及其杂质和降解产物的完全分离,从而提高了乙酰半胱氨酸及有关物质检测的灵敏度和检测的准确度。
50.具体地,乙酰半胱氨酸的有关物质可以包括l-胱氨酸、l-半胱氨酸、n,n-二乙酰-l-胱氨酸、n,s-二乙酰-l-半胱氨酸、(2r)-2-氨基-3-亚磺基丙酸一水合物、(r)-2-乙酰氨基-3-(((r)-2-氨基-2-羧乙基)二硫烷基)丙酸、n-(2-巯基乙基)乙酰胺等中的至少之一。为了表述方便,下表1中列出了乙酰半胱氨酸及各有关物质的化学结构式、化学名和本文中的代称。
51.表1乙酰半胱氨酸及相关物质列表
52.[0053][0054]
在一些具体的实施例中,色谱柱可以选用ace comixsil acrp色谱柱。
[0055]
进一步地,色谱柱的规格可以为250mm
×
4.6mm,直径为5μm。
[0056]
在一些具体的实施例中,作为流动相a的磷酸水溶液(即水-磷酸混合溶液)中水与磷酸的体积比优选为100:(0.01-0.05),例如,100:0.02、100:0.03、100:0.04。更优选地,流动相a中水与磷酸的体积比为100:0.03。
[0057]
另外,在一些具体的实施例中,作为流动相b的磷酸乙腈溶液(即乙腈-磷酸混合溶液)中乙腈与磷酸的体积比优选为100:(0.01-0.05),例如,100:0.02、100:0.03、100:0.04。更优选地,流动相b中乙腈与磷酸的体积比为100:0.03。
[0058]
本发明实施例提供的乙酰半胱氨酸及其有关物质的检测方法中,采用的色谱柱的固定相为耐水的阴阳离子嵌入修饰烷基键合硅胶,并且,采用的流动相中有机相比例高,对色谱柱的损耗小,可以延长色谱柱的使用寿命,从而降低检测成本。
[0059]
在一些实施例中,在进行梯度洗脱程序时,可根据下表2中所示的条件进行。
[0060]
表2梯度洗脱条件
[0061][0062]
也即是说,在进行梯度洗脱时,从0分钟到20分钟流动相a的体积占比从95-100%逐渐变化为15-25%,流动相b的体积占比从0-5%逐渐变化为75-85%;从20分钟到22分钟流动相a的体积占比从15-25%逐渐变化为95-100%,流动相b的体积占比从75-85%逐渐变化为0-5%;自22分钟始保持流动相a的体积占比为95-100%,流动相b的体积占比为0-5%;自32分钟始保持流动相a的体积占比为95-100%,流动相b的体积占比为0-5%直至检测结束。
[0063]
在一些优选的实施例中,在进行梯度洗脱程序时,可根据下表3中所示的条件进行。
[0064]
表3梯度洗脱条件
[0065][0066]
也即是说,在进行梯度洗脱时,从0分钟到20分钟流动相a的体积占比从100%逐渐变化为20%,流动相b的体积占比从0%逐渐变化为80%;从20分钟到22分钟流动相a的体积占比从20%逐渐变化为100%,流动相b的体积占比从80%逐渐变化为0%;自22分钟始保持流动相a的体积占比为100%;自32分钟始保持流动相a的体积占比为100%直至检测结束。
[0067]
通过设置恰当的梯度洗脱条件,可进一步保证乙酰半胱氨酸及其有关物质的分离效果,从而进一步提高检测的灵敏度和准确度。
[0068]
在一些具体的实施例中,高效液相色谱的流速可以设置为0.8-1.2ml/min,例如0.9ml/min、1.0ml/min、1.1ml/min等。优选地,流速可设置为1.0ml/min。
[0069]
色谱柱的柱温可以设置为25-35℃,例如26℃、27℃、28℃、29℃、30℃、31℃、32℃、33℃、34℃。优选地,柱温可设置为30℃。
[0070]
高效液相色谱可采用紫外检测,检测波长可设置为220nm。
[0071]
为保证样品的稳定性,进样温度可设置为4-8℃,例如5℃、6℃、7℃,优选为5℃。
[0072]
进样体积可以设置为5-15μl,例如6μl、7μl、8μl、9μl、10μl、11μl、12μl、13μl、14μl,优选为10μl。
[0073]
以上介绍了本发明的乙酰半胱氨酸及其有关物质的检测方法中高效液相色谱分析的条件和参数,下面具体介绍该检测方法的执行步骤。
[0074]
图1示出了根据本发明一实施例的乙酰半胱氨酸及其有关物质的检测方法的流程示意图。参加图1所示,该检测方法可以包括以下步骤s1至步骤s5。
[0075]
步骤s1:将待检测的乙酰半胱氨酸样品溶解于盐酸中并以稀释剂稀释制成第一浓度的供试品溶液。
[0076]
步骤s2:将乙酰半胱氨酸对照品溶解于稀释剂中制成第二浓度的对照品溶液。
[0077]
步骤s3:精密称量乙酰胺胱氨酸和各有关物质适量,并共同溶解于稀释剂中制成含第三浓度的乙酰半胱氨酸和第四浓度的各有关物质的系统适用性溶液。
[0078]
步骤s4:取系统适用性溶液进样进行高效液相色谱分析,以确定乙酰半胱氨酸和各有关物质的保留时间。
[0079]
步骤s5:分别取供试品溶液和对照品溶液进样进行高效液相色谱分析,通过加校正因子的主成分外标法计算供试品溶液中乙酰半胱氨酸和各有关物质的含量。
[0080]
本实施例中,可按照前述的高效液相色谱分析的条件和参数进行步骤s4和s5中的高效液相色谱分析。
[0081]
另外,步骤s1、s2和s3并没有确定的前后顺序,它们的顺序可以互换,也可以同时进行。
[0082]
进一步地,为保证分离效果和检测精密度,第三浓度与第四浓度的比例可设置在150:1至250:1范围内,例如为160:1、170:1、180:1、190:1、200:1、210:1220:1、230:1、240:
1。优选地,第三浓度与第四浓度的比例可为200:1。
[0083]
在一个具体的实施例中,第三浓度与第四浓度可以分别具体设置为8mg/ml和40μg/ml。
[0084]
进一步地,第一浓度可以设置在第三浓度的
±
10%范围内。第二浓度可以设置在第四浓度的
±
10%范围内。例如第一浓度可以设置为与第三浓度相同的8mg/ml。第二浓度可以设置为与第四浓度相同的40μg/ml。
[0085]
下面通过特定的具体实施例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与效果。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0086]
以下实施例中所用的乙酰半胱氨酸和有关物质对照品中,除了杂质o外,其余杂质都可以商业获得。杂质o由本技术发明人自行制备。
[0087]
杂质o((r)-2-乙酰氨基-3-(((r)-2-氨基-2-羧乙基)二硫烷基)丙酸)的合成路线如下式(1)所示。
[0088][0089]
杂质o的制备过程如下:
[0090]
将乙酰半胱氨酸、naoh溶液、l-半胱氨酸、mno2加入至50ml三口瓶中,搅拌成黑色浊液。反应2h后体系变为灰色浊液。过滤,得淡黄色溶液。将滤液冻干,得黄色固体2.48g。过色谱柱分离得到白色固体500mg。该白色固体经过核磁、lcms(liquid chromatography-mass spectrometry,液相色谱-质谱法)、红外、紫外等检测,确定其结构符合杂质o特征,其中lcms纯度98.95%,m/z=283.34(m+h
+
),1h-nmr(500mhz,dmso-d6)δ8.20(s,1h),4.55(s,1h),3.92(s,1h),3.35(m,2h),2.847-3.01(m,2h),1.86(s,3h)。
[0091]
实施例1
[0092]
本实施例1中色谱条件如下:
[0093]
色谱柱:ace comixsil acrp,250mm
×
4.6mm,5μm;流动相a:水-磷酸溶液(体积比100:0.03),流动相b:乙腈-磷酸溶液(体积比100:0.03);进行梯度洗脱,梯度洗脱设置如下表4所示:
[0094]
表4梯度洗脱条件
[0095][0096]
流速:1ml/min;柱温:30℃;检测波长:220nm;进样温度:5℃,进样体积:10μl。
[0097]
检测方法与结果如下:
[0098]
(1)溶液配制
[0099]
取乙酰半胱氨酸样品(本实施例中取乙酰半胱氨酸对照品)适量,精密称定,加0.01mol/l盐酸溶解并定量稀释制成每1ml中约含8mg的溶液,作为供试品溶液;取乙酰半胱氨酸对照品适量,精密称定,加稀释剂溶解并定量稀释制成每1ml含40μg的溶液,作为对照品溶液。
[0100]
(2)专属性
[0101]
取乙酰半胱氨酸和杂质a、杂质b、杂质c、杂质d、杂质e、杂质o、杂质r各适量,精密称定,置于同一量瓶中,加稀释剂溶解并稀释制成每1ml中含乙酰半胱氨酸8mg及各杂质均约为40μg的溶液,作为系统适用性溶液。精密量取10μl系统适用性溶液注入高效液相色谱仪,记录色谱图,确定乙酰半胱氨酸和各杂质的保留时间。系统适用性溶液的分离检测结果参见图2,出峰顺序依次为:保留时间rt5.115为杂质r、rt5.856为杂质b、rt8.003为杂质o、rt9.724为乙酰半胱氨酸、rt10.949为杂质d、rt11.743为杂质a、rt13.689为杂质e、rt19.39为杂质c。
[0102]
(3)精密度
[0103]
精密量取上述供试品溶液和对照品溶液进样进行高效液相色谱分析,记录色谱图。按加校正因子的主成分外标法计算6份供试品溶液中各有关物质含量。6份供试品溶液中检出杂质c的含量均为0.02%,rsd为0%,小于3.0%。可见,本发明的检测方法符合高效液相色谱法对有关物质检测的要求。
[0104]
(4)线性与范围
[0105]
取乙酰半胱氨酸及其杂质对照品适量,加稀释剂溶解并稀释制成浓度均为0.4mg/ml的溶液,作为储备液。分别移取储备液1ml置20ml、2ml置25ml、2ml置20ml、4ml置20ml、3ml置10ml量瓶中,用稀释剂稀释至刻度,作为线性供试液。以峰面积对浓度进行线性回归,得线性方程(列于下表5中)。结果表明,乙酰半胱氨酸及其杂质在线性范围内具有良好的线性关系。
[0106]
表5线性测定结果
[0107]
名称浓度范围(μg/ml)线性方程相关系数(r)乙酰半胱氨酸2.3669~136.0474y=12.4316x+0.53851.0000杂质a4.0469~124.0637y=9.9467x+4.33130.9996杂质b3.4260~121.1071y=5.2925x-1.53000.9998杂质c1.7886~113.0090y=17.9722x+6.84921.0000杂质d0.9779~123.9312y=33.2357x+0.56270.9999杂质e0.6679~114.8666y=56.2419x-5.26361.0000杂质o1.5801~103.7548y=11.8090x+3.36071.0000杂质r1.3371~116.2986y=12.2550x-2.30950.9999
[0108]
(5)定量限与检测限
[0109]
取乙酰半胱氨酸及其杂质对照品适量,配制成一系列溶液,s/n≥10时,作为定量限溶液;s/n≥3时,作为检测限溶液。乙酰半胱氨酸及其杂质的定量限和检测限结果见表6。
[0110]
表6定量限和检测限结果
[0111]
名称检测限(μg/ml)信噪比(s/n)定量限(μg/ml)信噪比(s/n)乙酰半胱氨酸0.71015.62.366915.0杂质a1.21414.94.046913.7杂质b1.02784.73.426013.5杂质c0.53666.11.788614.4杂质d0.32584.70.977912.5杂质e0.20043.40.667912.2杂质o0.47404.91.580114.2杂质r0.40114.31.337113.2
[0112]
从以上检测结果可看出,在本实施例的色谱条件下,乙酰半胱氨酸及其杂质以及降解产物可以完全分离。本发明的方法专属性强、准确度好、精密度高、重复性好,符合药物质量研究标准的技术要求,所得结果稳定可靠。
[0113]
对比例1
[0114]
对比例1中采用欧洲药典的检测方法,色谱条件如下:
[0115]
色谱柱:hypersil ods,250mm
×
4.6mm,5μm;流动相:水-乙腈-磷酸(体积比为97:3:0.1);进行等度洗脱,等度洗脱时间为30min。
[0116]
流速:1ml/min;柱温:30℃;检测波长:220nm;进样温度:5℃,进样体积:10μl。
[0117]
取乙酰半胱氨酸和杂质a、杂质b、杂质c、杂质d、杂质e、杂质o、杂质r各适量,精密称定,置同一量瓶中,加稀释剂溶解并稀释制成每1ml中含乙酰半胱氨酸50μg及各杂质均约为10μg的溶液,作为系统适用性溶液。将系统适用性溶液进样,按照以上色谱条件进行高效液相色谱分析。系统适用性溶液的色谱图参见图3。
[0118]
由图3可看出,在该色谱条件下,出峰顺序依次为:保留时间rt1.961为杂质a与e、rt2.177为杂质b、rt2.503为杂质o、rt4.243为乙酰半胱氨酸、rt8.009为杂质c、rt9.204为杂质d、rt18.894为杂质r。可见,杂质a、e、b和o出峰很早,几乎无保留,杂质a与e未分离,这影响杂质的定量结果和定性判断。
[0119]
对比例2
[0120]
对比例2中色谱条件如下:
[0121]
色谱柱:supersil c18,250mm
×
4.6mm,5μm;流动相:(10mmol/l磷酸二氢钾,30mmol/l庚烷磺酸钠,磷酸调节ph值2.0)-甲醇(体积比90:10);进行等度洗脱,等度洗脱时间为30min。
[0122]
流速:1ml/min;柱温:40℃;检测波长:220nm;进样温度:5℃,进样体积:20μl。
[0123]
系统适用性溶液的制备与对比例1相同。将系统适用性溶液进样,按照以上色谱条件进行高效液相色谱分析。系统适用性溶液的色谱图参见图4。
[0124]
由图4可看出,在该色谱条件下,出峰顺序依次为:保留时间rt2.267为杂质e、rt4.769为乙酰半胱氨酸、rt5.534为杂质r、rt6.556为杂质c、rt8.706为杂质d、rt11.938为杂质b和o、rt23.656为杂质a。可见,杂质b和o未分离,影响杂质的定量结果和定性判断。
[0125]
对比例3
[0126]
对比例3的色谱条件如下:
[0127]
色谱柱:supersil c18,250mm
×
4.6mm,5μm;流动相:(10mmol/l磷酸二氢钾,
20mmol/l庚烷磺酸钠,磷酸调节ph值2.0)-甲醇(体积比90:10);进行等度洗脱,等度洗脱时间为30min。
[0128]
流速:1ml/min;柱温:40℃;检测波长:220nm;进样温度:5℃,进样体积:20μl。
[0129]
系统适用性溶液的制备与对比例1相同。将系统适用性溶液进样,按照以上色谱条件进行高效液相色谱分析。系统适用性溶液的色谱图参见图5。
[0130]
由图5可知,在该色谱条件下,出峰顺序依次为:保留时间rt2.261为杂质e、rt5.286为乙酰半胱氨酸、rt6.057为杂质r、rt8.219为杂质c、rt8.929为杂质b、rt10.319为杂质d和o、rt14.434为杂质a。可见,杂质d和o未分离,影响杂质的定量结果和定性判断。
[0131]
对比例4
[0132]
对比例4的色谱条件如下:
[0133]
色谱柱:ace comixsil acrp,250mm
×
4.6mm,5μm;流动相a:水-甲酸(体积比100:0.03),流动相b:乙腈-甲酸(体积比100:0.03);进行梯度洗脱,梯度洗脱设置如下表7:
[0134]
表7梯度洗脱条件
[0135][0136][0137]
流速:1ml/min;柱温:30℃;检测波长:220nm;进样温度:5℃,进样体积:10μl。
[0138]
系统适用性溶液的制备与实施例1相同。将系统适用性溶液进样,按照以上色谱条件进行高效液相色谱分析。系统适用性溶液的色谱图参见图6。
[0139]
由图6可知,在该色谱条件下,出峰顺序依次为:保留时间rt3.721为杂质a、rt18.327为乙酰半胱氨酸。仅乙酰半胱氨酸与杂质a出峰,其余杂质均无响应,影响杂质的定量结果和定性判断。
[0140]
在此处所提供的说明书中,说明了大量具体细节。然而,能够理解,本发明的实施例可以在没有这些具体细节的情况下实践。在一些实例中,并未详细示出公知的方法、结构和技术,以便不模糊对本说明书的理解。
[0141]
至此,本领域技术人员应认识到,虽然本文已详尽示出和描述了本发明的多个示例性实施例,但是,在不脱离本发明精神和范围的情况下,仍可根据本发明公开的内容直接确定或推导出符合本发明原理的许多其他变型或修改。因此,本发明的范围应被理解和认定为覆盖了所有这些其他变型或修改。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1