一种裸花紫珠胶囊中木犀草苷的含量测定方法与流程
1.本发明涉及裸花紫珠胶囊含量检测领域,特别涉及一种裸花紫珠胶囊中木犀草苷的含量测定方法。
背景技术:
2.裸花紫珠(学名:callicarpa nudiflora hook.et arn.)是马鞭草科,紫珠属灌木至小乔木,高可达7米;老枝无毛而皮孔明显,小枝、叶柄与花序密生灰褐色分枝茸毛。叶片卵状长椭圆形至披针形,表面深绿色,干后变黑色,聚伞花序开展,苞片线形或披针形;花萼杯状,花冠紫色或粉红色,花药椭圆形,细小,果实近球形,红色。
3.裸花紫珠叶药用,有止血止痛、散瘀消肿之效。治外伤出血、跌打肿痛、风湿肿痛、肺结核咯血、胃肠出血等功效。
4.目前市场上裸花紫珠制剂主要有颗粒剂、胶囊、片剂、分散片、合剂、软胶囊、栓剂等。不同的制剂类型,所使用的辅料并不相同,因此本发明提出一种裸花紫珠胶囊中木犀草苷的含量测定方法。
技术实现要素:
5.鉴以此,本发明提出一种裸花紫珠胶囊中木犀草苷的含量测定方法,解决上述问题。
6.本发明的技术方案是这样实现的:
7.一种裸花紫珠胶囊中木犀草苷的含量测定方法,采用高效液相色谱仪进行检测,所述高效液相色谱条件为:
8.色谱柱:c18;
9.流速:0.8-1.2ml/min;
10.检测波长:340-360nm;
11.流动相a:高氯酸钠缓冲盐溶液
12.流动相b:甲醇-乙腈混合液
13.柱温:25-35℃;
14.进样体积:10-30μl;
15.洗脱梯度程序:
16.时间(min)a%(v/v)b%(v/v)0-15.075-8525-1515.01-35.055-6545-3535.1-65.070-7530-2565.1-7080-8520-15
17.溶剂:60-80%v/v甲醇溶液。
18.进一步的,所述流动相a高氯酸钠缓冲盐溶液浓度为0.5-0.8mol/l,使用磷酸溶液
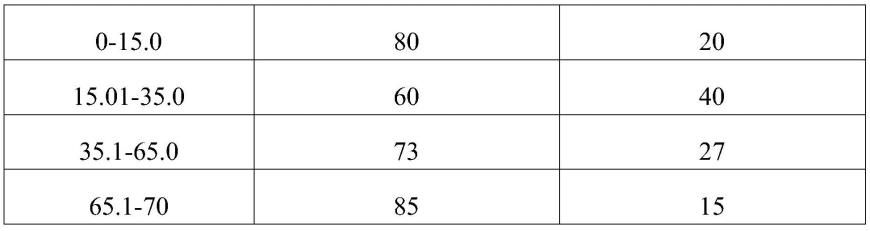
调节ph值至3-4。
19.进一步的,所述流动相b中甲醇和乙腈体积比为3-4:1.5-2.5。
20.进一步的,所述色谱柱c18规格为3.0
×
150mm,填料粒径为2μm。
21.进一步的,所述洗脱梯度参数为:
22.时间(min)a%(v/v)b%(v/v)0-15.0802015.01-35.0604035.1-65.0732765.1-708515
23.进一步的,所述磷酸溶液浓度为0.1-1mol/l。
24.进一步的,所述检测波长为350nm。
25.进一步的,所述对照品溶液制备方法为称取木犀草苷至容量瓶,加溶剂溶解稀释至浓度为0.1-1mg/ml。
26.进一步的,所述供试品溶液的制备方法包括以下步骤:
27.(1)取一颗裸花紫珠胶囊内容物粉碎,将粉碎后裸花紫珠胶囊内容物转移至20ml容量瓶中,加8-10ml的质量浓度为50-60%乙醇溶液超声5-8min,超声温度为30-40℃,超声功率为80-120w,超声后的裸花紫珠胶囊内容物溶液加0.5-1.5ml的盐酸溶液和甲醇补足至刻度线,所述盐酸溶液摩尔浓度为1-2mol/l,制得粗提液1;
28.(2)将粗提液1进行超声50-70min,超声功率为50-70w,超声温度为50-60℃,超声后放冷,过滤,制得供试品溶液。
29.进一步的,所述盐酸溶液摩尔浓度为1.5mol/l。
30.与现有技术相比,本发明的有益效果是:
31.本发明中使用高氯酸钠缓冲盐溶液为流动相a,使用甲醇乙腈混合液为流动相b,合理设置洗脱梯度,调整不同时段中有机相的占比能够避开辅料以及裸花紫珠中其他组分对木犀草苷峰的干扰。进一步准确有效检测裸花紫珠胶囊中木犀草苷的含量。
32.本发明中供试品溶液先将裸花紫珠胶囊内容物粉碎,加入乙醇溶液超声,超声后再添加盐酸溶液和甲醇,混匀,超声。提高供试品溶液的洗脱强度,样品的溶剂带在色谱柱中快速迁移,同时也避免样品在洗脱过程中溶解于流动相中,造成木犀草苷峰宽过宽或劈叉等现象出现。随之引起柱效降低等一系列问题。
附图说明
33.图1实施例1线性关系试验标准曲线
34.图2实施例1木犀草苷对照品溶液色谱图
35.图3实施例1裸花紫珠胶囊供试品溶液色谱图
具体实施方式
36.为了更好理解本发明技术内容,下面提供具体实施例,对本发明做进一步的说明。
37.本发明实施例所用的实验方法如无特殊说明,均为常规方法。
38.本发明实施例所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
39.本发明木犀草苷对照品来源于中国药品生物制品检定所,裸花紫珠胶囊批号为20160701。
40.实施例1
41.色谱柱:inertsustain c18(3.0
×
150mm,2μm);
42.流速:1ml/min;
43.检测波长:350nm;
44.流动相a:高氯酸钠缓冲盐溶液浓度为0.7mol/l,使用磷酸溶液调节ph值至3.6;
45.流动相b:甲醇和乙腈体积比为3.6:2;
46.柱温:30℃;
47.进样体积:20μl;
48.洗脱梯度程序:
49.时间(min)a%(v/v)b%(v/v)0-15.0802015.01-35.0604035.1-65.0732765.1-708515
50.实施例2
51.色谱柱:inertsustain c18(3.0
×
150mm,2μm);
52.流速:0.8ml/min;
53.检测波长:340nm;
54.流动相a:高氯酸钠缓冲盐溶液浓度为0.5mol/l,使用磷酸溶液调节ph值至3;
55.流动相b:甲醇和乙腈体积比为3:1.5;
56.柱温:25℃;
57.进样体积:20μl;
58.洗脱梯度程序:
59.时间(min)a%(v/v)b%(v/v)0-15.0752515.01-35.0554535.1-65.0703065.1-708020
60.实施例3
61.色谱柱:inertsustain c18(3.0
×
150mm,2μm);
62.流速:1.2ml/min;
63.检测波长:360nm;
64.流动相a:高氯酸钠缓冲盐溶液浓度为0.8mol/l,使用磷酸溶液调节ph值至4;
65.流动相b:甲醇和乙腈体积比为4:2.5;
66.柱温:35℃;
67.进样体积:20μl;
68.洗脱梯度程序:
69.时间(min)a%(v/v)b%(v/v)0-15.0851515.01-35.0653535.1-65.0752565.1-708515
70.试验例1
71.选用实施例1的色谱条件进行含量方法学检验,检验项目有精密度试验、准确度试验、线性关系试验、稳定性试验、系统适用性试验。
72.1、线性关系试验
73.储备液a:取木犀草苷对照品约50mg,精密称定,置50ml容量瓶中,加70%v/v甲醇适量充分使溶解并定量至刻度。
74.线性关系试验溶液:将储备液a加70%v/v甲醇制备成50%、75%、100%、125%、150%线性关系溶液,其中100%线性关系溶液浓度为0.1mg/ml。
75.以峰面积为横坐标、浓度为纵坐标绘制线性回归方程。
76.参见图1,线性回归方程为y=909535x-272,r2=1。
77.实验结果表明木犀草苷对照品在50%-150%范围内线性关系良好。
78.2、精密度试验
79.对照品溶液的配制:取木犀草苷对照品约10mg,精密称定,置10ml容量瓶中,加70%甲醇适量充分使溶解并定量至刻度,制得对照品溶液,进样六次,计算主峰保留时间和峰面积rsd值。
80.名称主峰保留时间峰面积rsd(%)0.090.13
81.实验结果表明,本发明的色谱条件精密度符合要求。
82.3、回收率试验
83.配制浓度为0.1mg/ml的对照品溶液,以对照品加入量与所取供试品中测定成分之比为(1.5:1,1:1,0.5:1)每种浓度分别制备3份供试品溶液进行测定,用9份样品的测定结果进行验证准确度;通过计算溶液测定值与理论值的比值测定准确度回收率。
84.[0085][0086]
实验结果表明,木犀草苷平均回收率为99.90%,rsd为0.22%,回收率符合要求。
[0087]
4、溶液稳定性试验
[0088]
对照品溶液的配制:取木犀草苷对照品约10mg,精密称定,置100ml容量瓶中,加70%v/v甲醇适量充分使溶解并定量至刻度,制得对照品溶液。
[0089]
供试品溶液:取一颗裸花紫珠胶囊内容物粉碎,将粉碎后裸花紫珠胶囊内容物转移至20ml容量瓶中,加8-10ml的质量浓度为55%乙醇溶液超声7min,超声温度为35℃,超声功率为100w,超声后的裸花紫珠胶囊内容物溶液加1ml摩尔浓度为1.5mol/l的盐酸溶液和甲醇滴定至刻度线,制得粗提液1;
[0090]
将粗提液1使用超声处理60min,超声功率为60w,超声温度为55℃,超声后放冷,过滤,制得供试品溶液。
[0091]
取对照品溶液和供试品溶液于0、2、4、6、8、10、12、18、24h进样分析,计算保留时间和峰面积的rsd值。
[0092]
名称保留时间rsd(%)峰面积rsd(%)对照品溶液0.290.35供试品溶液0.240.30
[0093]
实验结果表明,溶液稳定性试验rsd(%)小于2.0%,对照品溶液和供试品溶液在24小时内稳定。
[0094]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1