一种内消瘰疬片的质量检测方法与流程
1.本发明属于中药制剂检测技术领域,具体地说,涉及一种内消瘰疬片的质量检测方法。
背景技术:
2.内消瘰疬片是由玄参、牡蛎(煅)、夏枯草、天花粉、党参、黄芪、白芥子、三棱、莪术、赤芍、浙贝母、海藻、穿山甲(炮)、羊乳、地龙15味药材制成的成方制剂,是南京市中西医结合医院的医院制剂(苏药制字z04000312),与《中国药典》收载的配方不同。临床上内消瘰疬片与抗结核一线治疗药物异烟肼等联合用药治疗肺结核及耐多药结核,降低肝功能损伤和胃肠道反应大副作用;西药联合内消瘰疬丸治疗颈部淋巴结结核具有提高临床疗效,提高杀菌效果并改善预后;内消瘰疬丸联合左甲状腺素钠等药物治疗良性甲状腺结节具有疗效,减少西药不良反应,增强患者治疗依从性。
3.现有技术采用hplc测定迷迭香酸控制内消瘰疬片质量,应用rp-hplc方法测定其中柚皮苷含量,采用hplc测定处方中夏枯草迷迭香酸及玄参哈巴俄苷的含量,现有hplc质量控制方法集中在夏枯草中迷迭香酸、玄参中哈巴俄苷成分,未有hplc指纹图谱研究及多指标成分定量分析。本发明拟建立内消瘰疬片hplc指纹图谱方法和多成分含量分析,拟开展4个主要成分(芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、迷迭香酸)同时含量测定,形成指纹图谱和多成分含量测定二者“模糊”和“精准”质量联合控制,进一步提升内消瘰疬片质量控制标准,可以为评价和控制其质量提供参考依据。
技术实现要素:
4.发明目的:本发明的目的是为了克服现有技术的不足,开发一种关于内消瘰疬片的质量检测方法,通过本发明的方法可以客观,正确,有效的控制内消瘰疬片的质量,对确保其安全性与有效性,保证临床质量具有重要的指导意义。
5.技术方案:为实现以上目的,本发明采用的技术方案为:
6.一种内消瘰疬片的质量检测方法,它包括以下步骤:
7.步骤1:内消瘰疬片供试品溶液的制备
8.取不同批次的内消瘰疬片适量,打粉,粉末过筛,取粉末适量,精密加入甲醇,超声处理,滤过,取续滤液,至水浴锅蒸发皿蒸干,残渣加甲醇溶解,置于容量瓶,用甲醇定容至刻度线,摇匀,离心,经0.45μm微孔滤膜滤过,即得;
9.步骤2:混合对照品溶液的制备
10.分别精密称取对羟基苯甲酸、芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、异槲皮素、迷迭香酸、肉桂酸、槲皮素、芒柄花素对照品,制得单个对照品溶液;精密吸取上述9种单个对照品,置于容量瓶中,加甲醇定容至刻度线,过0.45μm微孔滤膜,即得混合对照品,备用;
11.步骤3、色谱图测定
12.分别吸取步骤1的供试品溶液和步骤2的混合对照品溶液,注入高效液相色谱仪,
得到供试品溶液的色谱图和混合对照品溶液的色谱图;
13.步骤4、将步骤3中获得的内消瘰疬片供试品溶液的指纹图谱导出,并导入中药色谱指纹图谱相似度评价系统2004a版;选择不同批次内消瘰疬片供试品溶液的色谱图中均存在的色谱峰作为共有峰,建立内消瘰疬片供试品溶液的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;并根据混合对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分。
14.作为优选方案,以上所述的一种内消瘰疬片的质量检测方法,步骤1、内消瘰疬片供试品溶液的制备方法为:
15.取不同批次的内消瘰疬片研磨成粉,粉末过60目筛,取粉末1g,置于50ml磨口具塞锥形瓶中,精密加入甲醇30ml,密封,超声处理30min,滤过,取续滤液,至水浴锅70℃蒸发皿蒸干,残渣加甲醇溶解,置于5ml容量瓶,用甲醇定容至刻度线,摇匀,12000r
·
min-1
离心10min,经0.45μm微孔滤膜滤过,即得。
16.作为优选方案,以上所述的一种内消瘰疬片的质量检测方法,步骤2混合对照品溶液的制备方法为:
17.精密称取对羟基苯甲酸、芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、异槲皮素、迷迭香酸、肉桂酸、槲皮素、芒柄花素对照品,制得浓度分别为0.882g
·
l-1
、0.885g
·
l-1
、0.880g
·
l-1
、1.442g
·
l-1
、0.886g
·
l-1
、1.122g
·
l-1
、2.414g
·
l-1
、0.703g
·
l-1
、0.126g
·
l-1
的单个对照品溶液;精密吸取上述9种单个对照品溶液各1ml,置于20ml容量瓶中,加甲醇定容至刻度线,过0.45μm微孔滤膜,即得混合对照品,备用。
18.作为优选方案,以上所述的一种内消瘰疬片的质量检测方法,步骤3的色谱条件为ymc-pack ods-ac
18
色谱柱,规格为4.6mm
×
250mm,5μm;流动相:a相为乙腈-b相为0.1%磷酸水溶液,梯度洗脱;流速:1.0ml
·
min-1
;柱温35℃;检测波长:230nm;进样量:10μl。
19.作为优选方案,以上所述的一种内消瘰疬片的质量检测方法,步骤3的梯度洗脱程序为:0~30min,5%a;30~105min,5%~30%a;105~175min,30%~100%a;175~180min,100%~5%a;180~185min,5%a。
20.本发明所述的一种内消瘰疬片的质量检测方法,步骤5对照指纹图谱共有14个共有峰,其中对羟基苯甲酸为1号峰,保留时间为24.415min、芍药苷为4号峰,保留时间为61.410min、苯甲酸为5号峰,保留时间为66.719min、毛蕊异黄酮葡萄糖苷为6号峰,保留时间为71.745min、异槲皮素为7号峰,保留时间为74.833min、迷迭香酸为9号峰,保留时间为85.991min、肉桂酸为11号峰,保留时间为95.064min、槲皮素为12号峰,保留时间为98.040min、芒柄花素为13号峰,保留时间为98.040min。
21.指纹图谱检测条件的优化
22.1、检测波长的选择
23.对供试品溶液进行全波长扫描,对比波长为210nm、230nm、254nm、280nm、330nm、350nm下的色谱图,发现目标成分在230nm左右均有较好吸收,峰形较好。选择230nm为检测波长,(如图5和图6不同波长的色谱图)。
24.2、流动相的选择
25.本研究考察不同流动相(乙腈-0.2%磷酸水溶液,甲醇-0.1%磷酸水溶液,乙腈-0.1%甲酸水溶液),结果采用乙腈-0.1%磷酸水系统,分离效果及峰形良好,基线较为平
稳,故选择乙腈-0.1%磷酸水作为流动相。
26.3、梯度洗脱方式的筛选
27.本发明筛选了以下4个不同的梯度洗脱方式,最终得到本发明最佳的梯度洗脱方式。
28.洗脱梯度一:色谱图如图7。
[0029][0030][0031]
洗脱梯度二:色谱图如图8。
[0032]
时间(min)乙腈体积浓度0.1%磷酸水溶液体积浓度05%95%305%95%10530%70%130100%0%1355%95%1405%95%
[0033]
洗脱梯度三:色谱图如图9。
[0034]
时间(min)乙腈体积浓度0.1%磷酸水溶液体积浓度05%95%305%95%10530%70%150100%0%1555%95%1605%95%
[0035]
洗脱梯度四:色谱图如图10。
[0036]
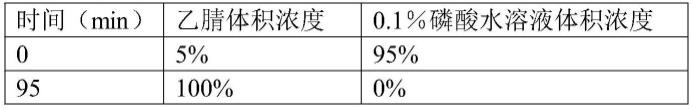
时间(min)乙腈体积浓度0.1%磷酸水溶液体积浓度05%95%305%95%10530%70%175100%0%1805%95%1855%95%
[0037]
其中以洗脱梯度四最佳,因此,本发明优选洗脱梯度四为本发明的洗脱条件。
[0038]
3、柱温及流速的选择
[0039]
本研究考察不同的柱温(25,30,35℃)以及流速(0.6、0.8、1.0ml
·
min-1
对色谱峰的影响,结果表明在230nm波长下,柱温为35℃、流速为1.0ml
·
min-1
,所得的色谱峰峰形、数
目及分离度等均表现良好。
[0040]
本发明的有益效果是:
[0041]
(1)本发明筛选出最佳的供试品与对照品的制备方法,通过大量实验筛选出最佳的流动相组成和梯度洗脱方式等色谱条件,建立的内消瘰疬片指纹图谱和含量测定方法,不但能有效地表征内消瘰疬片的质量,有利于全面监控内消瘰疬片的质量。
[0042]
(2)本发明方法具有稳定性好、精密度高、准确度高、重现性好等的优点。可以全面、客观准确的评价和控制内消瘰疬片的质量。
附图说明
[0043]
图1为供试品溶液的hplc色谱图。
[0044]
图2为混合对照品hplc色谱图。
[0045]
图3为10批不同批次内消瘰疬片hplc指纹图谱。
[0046]
图4为芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷和迷迭香酸对照品hplc色谱图。
[0047]
图5为210nm、230nm、254nm不同波长下的色谱图。
[0048]
图6为280nm、330nm、350nm不同波长下的色谱图。
[0049]
图7洗脱梯度一的色谱图。
[0050]
图8洗脱梯度二的色谱图。
[0051]
图9洗脱梯度三的色谱图。
[0052]
图10洗脱梯度四的色谱图。
具体实施方式
[0053]
下面将结合实施例对本发明的实施方案进行详细描述,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0054]
1仪器和试药
[0055]
1.1仪器lc-20at型高效液相色谱仪(spd-m20a二极管阵列检测器)(日本岛津公司);bp211d型万分之一精确分析天平(北京赛多利斯仪器系统有限公司);fa1004n型十万分之一精确电子分析天平(北京赛多利斯仪器系统有限公司);hh-6型数字显视恒温水浴锅(常州市江南实验仪器厂);shz-d(iii)型循环水式真空泵(巩义市予华仪器有限责任公司);kq5200de型数控超声波清洗器(频率:40khz,昆山市超声仪器有限公司)。
[0056]
1.2试药芍药苷(批号ps000825,含量》98%)购自成都普思生物科技有限公司;肉桂酸(批号180607,含量》98%)购自南京森贝伽生物科技有限公司);对羟基苯甲酸(批号:100278-201906,含量》98%)、苯甲酸(批号:100419-200301,含量》98%)、毛蕊异黄酮葡萄糖苷(批号111902-201102,含量》97.9%)、异槲皮苷(批号111809-201804,含量》98%)、迷迭香酸(批号111871-201611,含量》90.5%)、槲皮素(批号100081-201509,含量》98.1%)、芒柄花素(批号111703-200602,含量》98%)购自中国食品药品检定研究院。乙腈为色谱纯,水为纯化水,其余试剂均为分析纯。内消瘰疬片10批(南京市中西医结合医院自制,批准文号:苏药制字z04000312,批号09180104、09180304、09180601、09180709、09180903、09181104、09180903、09190104、09190203、200301)。
[0057]
实施例1
[0058]
1、一种内消瘰疬片的质量检测方法,它包括以下步骤:
[0059]
步骤1:内消瘰疬片供试品溶液的制备
[0060]
取以上10批次的内消瘰疬片样品适量,打粉,粉末过60目筛,取1g,置于50ml磨口具塞锥形瓶中,精密加入甲醇30ml,密封,超声处理30min,滤过,取续滤液,至水浴锅70℃蒸发皿蒸干,残渣加甲醇溶解,置于5ml容量瓶,用甲醇定容至刻度线,摇匀,离心(12000r
·
min-1
,10min),经0.45μm微孔滤膜滤过,即得。
[0061]
步骤2:对照品溶液的制备
[0062]
精密称取对羟基苯甲酸、芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、异槲皮素、迷迭香酸、肉桂酸、槲皮素、芒柄花素对照品,制得浓度为0.882g
·
l-1
、0.885g
·
l-1
、0.880g
·
l-1
、1.442g
·
l-1
、0.886g
·
l-1
、1.122g
·
l-1
、2.414g
·
l-1
、0.703g
·
l-1
、0.126g
·
l-1
对照品溶液。精密吸取上述9种对照品各1ml,置于20ml容量瓶中,加甲醇定容至刻度线,过0.45μm微孔滤膜,即得混合对照品。冷藏,备用。
[0063]
步骤3、色谱图测定
[0064]
分别吸取步骤1的供试品溶液和步骤2的混合对照品溶液,注入高效液相色谱仪,得到供试品溶液的色谱图和混合对照品溶液的色谱图;
[0065]
色谱条件为:ymc-pack ods-a c18色谱柱(4.6mm
×
250mm,5μm);流动相:乙腈(a)-0.1%磷酸水溶液(b),梯度洗脱(0~30min,5%a;30~105min,5%~30%a;105~175min,30%~100%a;175~180min,100%~5%a;180~185min,5%a);流速:1.0ml
·
min-1
;柱温35℃;检测波长:230nm;进样量:10μl。
[0066]
步骤4、将步骤3中获得的内消瘰疬片供试品溶液的指纹图谱导出,并导入中药色谱指纹图谱相似度评价系统2004a版;以中位数法对样品的指纹图谱进行峰点校正,数据匹配分析,生成共有模式图,建立内消瘰疬片供试品溶液的对照指纹图谱,计算各共有峰的相对保留时间和相对峰面积;建立的各批次内消瘰疬片与共有模式间的相似度如下表1所示:
[0067]
表1各批次样品与共有模式间的相似度
[0068]
[0069]
本发明利用相似度软件对10批内消瘰疬片指纹图谱进行分析,各批样品相似度均在0.66以上,各批内消瘰疬片相似度良好。
[0070]
并根据混合对照品溶液色谱图的保留时间标注对照指纹图谱中峰的化学成分。指纹图谱共有14个共有峰,其中对羟基苯甲酸为1号峰,保留时间为24.415min、芍药苷为4号峰,保留时间为61.410min、苯甲酸为5号峰,保留时间为66.719min、毛蕊异黄酮葡萄糖苷为6号峰,保留时间为71.745min、异槲皮素为7号峰,保留时间为74.833min、迷迭香酸为9号峰,保留时间为85.991min、肉桂酸为11号峰,保留时间为95.064min、槲皮素为12号峰,保留时间为98.040min、芒柄花素为13号峰,保留时间为98.040min。
[0071]
2、hplc指纹图谱方法学考察
[0072]
1、精密度试验选取内消瘰疬片样品(批号09190203),按以上步骤1方法配制供试品溶液,按照以上步骤3的色谱条件,连续进样6次,每次进样量10μl,以芍药苷为参考峰,记录14个共有峰的保留时间和峰面积。各共有峰保留时间rsd均小于1.5%,峰面积的rsd均小于3%,结果表明仪器精密度良好。
[0073]
2、稳定性试验选取内消瘰疬片样品(批号09190203),按以上步骤1方法配制供试品溶液,按以上步骤3的色谱条件进样检测,分别与0、4、8、16、24h重复进样,每次进样量10μl,以芍药苷为参考峰,记录14个共有峰的保留时间以及峰面积。各共有峰保留时间rsd均小于0.7%,峰面积的rsd均小于3%,结果表明供试品在24h内稳定性良好。
[0074]
3、重复性试验选取内消瘰疬片样品(批号09190203)6份,按步骤1方法配制供试品溶液,按以上步骤3的色谱条件进样检测,每次进样量10μl,以芍药苷为参考峰,记录14个共有峰的保留时间以及峰面积。各成分的保留时间rsd均小于0.36%、峰面积的rsd均小于3%,结果表明本方法的重复性良好。
[0075]
实施例2内消瘰疬片中多指标成分定量分析
[0076]
1、线性关系考察
[0077]
精密称取芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、迷迭香酸对照品,制得浓度分别为0.269g
·
l-1
、0.039g
·
l-1
、0.013g
·
l-1
、0.034g
·
l-1
对照品混合溶液。
[0078]
按照实施例1的步骤3的色谱条件测定,进样体积为2μl、4μl、8μl、12μl、16μl、20μl。芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷和迷迭香酸对照品hplc色谱图如图4。分别以对照品的峰面积(y)和进样量(x)建立4种成分的线性方程如表2,结果表明,4个成分的峰面积与进样量之间呈现良好的线性关系。
[0079]
表2标准曲线
[0080]
对照品名称线性方程r线性范围(μg)芍药苷y=249635x+216970.99960.134~2.69苯甲酸y=158962x+242950.99960.019~0.39毛蕊异黄酮葡萄糖苷y=27972x+5035.10.99960.006~0.13迷迭香酸y=53944x-639500.99950.017~0.34
[0081]
2、含量测定取以上10批内消瘰疬片样品,按实施例1步骤1相同的方法制备得到10批次样品的供试品溶液,按照实施例1的步骤3的色谱条件进样测定,记录各供试品溶液的峰面积,代入以上标准曲线方程,计算各成分的含量,具体结果见表3。
[0082]
表3内消瘰疬片样品中4种成分含量测定结果(n=10)
[0083][0084][0085]
3、方法学考察
[0086]
3.1精密度试验
[0087]
选取内消瘰疬片样品(批号09190203),按以上实施例1的步骤1方法配制供试品溶液,按以上实施例1步骤3的色谱条件进样检测,连续进样6次,每次进样量10μl,记录色谱图。结果芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、迷迭香酸峰面积rsd分别为0.60%、0.86%、0.75%、0.49%,表明仪器精密度良好。
[0088]
3.2稳定性试验
[0089]
选取内消瘰疬片样品(批号09190203),按以上实施例1的步骤1方法配制供试品溶液,按以上实施例1步骤3的色谱条件进样检测,分别与0、4、8、16、24h重复进样,每次进样量10μl,记录色谱图。结果芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、迷迭香酸的峰面积rsd分别为0.86%、0.37%、1.04%、0.46%,表明供试品在24h内稳定性良好。
[0090]
3.3重复性试验
[0091]
选取内消瘰疬片样品(批号09190203)6份,按以上实施例1的步骤1方法配制供试品溶液,按以上实施例1步骤3的色谱条件进样检测,每次进样量10μl,记录色谱图,结果芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、迷迭香酸峰面积rsd分别为2.84%、2.99%、2.65%、1.64%,表明本方法的重复性良好。
[0092]
3.4加样回收率试验
[0093]
选取内消瘰疬片样品(批号09180104),精密称取1g,共9份,置于50ml锥形瓶中,按以上实施例1的步骤1方法配制供试品溶液,在最后固体物加按样品中芍药苷含量的80%、100%、120%混合对照品溶解,置于5ml容量瓶,用甲醇定容至刻度线。按以上实施例1步骤3的色谱条件进样检测,测定其hplc色谱图,分别记录4种成分的峰面积,根据以上表2的标准曲线计算4种成分的含量和加样回收率,计算4种成分平均回收率分别为99.76%、98.87%、97.37%、98.91%,rsd分别为2.72%、1.57%、2.58%、1.75%。芍药苷、苯甲酸、毛蕊异黄酮葡萄糖苷、迷迭香酸4个成分回收率测定结果见表4。
[0094]
表4回收率测定结果(n=9)
[0095]
[0096][0097]
由以上实验结果表明,本发明建立的内消瘰疬片指纹图谱检测方法和含量测定方法,精密度、稳定性和重复性好,能有效地表征内消瘰疬片的质量,有利于全面监控其质量。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1