一种大规模标注lncRNA功能的方法与流程
本发明属于生物信息学领域,特别涉及一种大规模标注lncRNA功能的方法。
背景技术:
长链非编码RNA(long non-coding RNA,lncRNA)是一种重要的非编码RNA,它在真核生物中被广泛转录。一般,lncRNA具有低的表达水平,中等的序列保守性,和高的组织特异性。越来越多的生物实验已经证实lncRNA能在细胞中发挥广泛而又重要的作用,比如基因调控、剪接控制、以及X染色体剂量补偿等。lncRNA还和人类疾病的发生、发展和防治都有着密切联系。因此,确定lncRNA的功能对于揭示其在生理及病理过程中的作用机制、疾病诊断和防治都有重要的意义,但是,目前人们仅仅对很少量的lncRNA的功能了解比较充分。最近,预测和识别lncRNA功能的研究引起了越来越多研究者的兴趣。
确定lncRNA的功能,在生物学领域,一般采取非编码RNA沉默和定位分析、RNA结合蛋白免疫沉淀技术、紫外交联免疫沉淀、环状染色质构象捕获、RNA反义纯化、RNA纯化的染色质分离和捕获杂交分析RNA靶点等,尽管这些技术能在一定程度上识别lncRNA的部分功能,但是由于实验设计复杂、代价高昂,而lncRNA的功能具有多样化和特异性强的特点,难以大规模应用于lncRNA功能识别。随着微阵列和新一代测序等高通量技术的发展,获得了大量与lncRNA有关的生物数据(lncRNA序列、表达谱、与蛋白质的相互作用等),这为从计算上预测lncRNA的功能提供了条件。
近年来,已有一些研究者利用这些生物数据预测lncRNA的功能,比如,Guttman等人在4种小鼠细胞种通过基因组范围染色质状态谱发现了大约1600种lncRNA,并开发了一种方法进行lncRNA功能预测;Liao等人根据公开的微阵列表达谱数据,通过构造编码-非编码基因共表达网络标注了340个lncRNA的可能功能;Cabili与他的合作者编制了一个包含8000多种人类lincRNA的参考目录,并通过编码基因和非编码基因的共表达信息对它们进行了功能标注。这些方法基本上都是基于基因表达谱和一些局部信息,所以仅仅少量的lncRNA的功能可以被推断出来。近几年来,也出现了结合其它信息进行lncRNA功能标注的方法,例如,lncRNA2Function等。
技术实现要素:
本发明提供一种大规模标注lncRNA功能的方法,其可以一次对大量lncRNA的功能进行标注,大大降低lncRNA功能标注的成本,降低费用。
本发明的技术方案如下:
1)根据lncRNA与蛋白质的共表达数据、相互作用数据计算lncRNA和蛋白质的皮尔逊相关系数,并根据相关系数构造lncRNA-蛋白质关系网络。2)根据lncRNA在人类24个组织或者细胞类型中的表达谱计算lncRNA之间的皮尔逊相关系数,据此构造lncRNA相似性网络。3)根据蛋白质相互作用数据构造蛋白质相互作用网络,并结合lncRNA-蛋白质和lncRNA相似性网络构建全局网络。4)利用上述构建的全局网络,根据公式(1)计算lncRNA节点和蛋白质节点的Katz度量,此Katz度量代表lncRNA节点和蛋白质节点的相似性度量,Katz度量值越大,说明这个蛋白质与lncRNA越相似。5)对上述步骤得到的lncRNA和蛋白质相似矩阵进行降序排列,按照分值选择其中前N个蛋白质,分别找出前N个蛋白质中每个蛋白质所对应的功能注释,对每个功能注释,根据公式(2)计算此lncRNA具有该功能的概率。
本发明与现有标注lncRNA功能的方法相比,现有的大部分方法都是基于基因的表达谱和基因的一些局部信息,因此一次仅能对少量的lncRNA进行功能注释,而本发明是根据全局网络进行计算推断的,所以一次可以对全基因组的lncRNA进行功能注释。此外,本发明不但考虑了基因表达谱信息,也结合了lncRNA与蛋白质的相互作用信息以及蛋白质之间的相互作用信息。与现有的方法相比,本发明利用了更多的生物数据,可以显著地提高lncRNA功能预测的准确度,同时,本发明可以一次对大量lncRNA进行功能预测,有效的解决了现有计算方法的问题,也为生物实验进行lncRNA功能注释提供了有价值的参考。
附图说明
图1是本发明实施例整个过程的处理流程示意图。
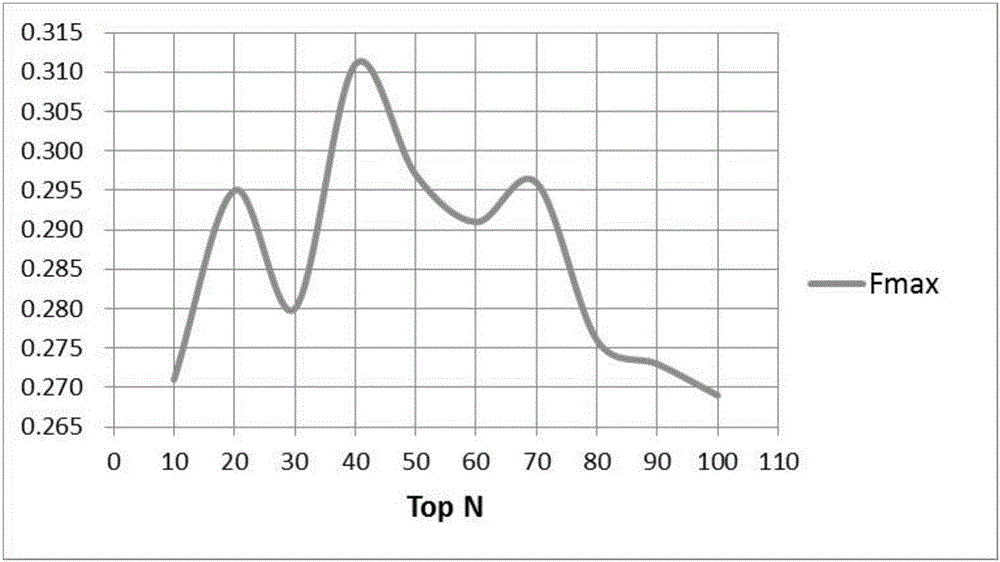
图2为本发明实施例N取不同值时的性能变化曲线图,当N选择不同的值时,Fmax的值波动较大,最好的性能(Fmax最大)出现在N近似是40时。
图3本发明实施例网络中包含或去掉PPI时的准确率-召回率曲线图。
图4在手工标注的55个lncRNA上,本发明实施例和LncRNA2Function分别正确注释的lncRNA的个数比较示意图。
图5在全基因组上,本发明实施例和LncRNA2Function分别正确注释的lncRNA的个数比较示意图。
图6在不同GO深度下,本发明实施例和LncRNA2Function分别注释lncRNA的个数比较示意图。
具体实施方式
下面将结合附图和实施例对本发明做进一步详细说明。
本发明的原理是:根据lncRNA-蛋白质的共表达数据及相互作用数据、lncRNA的表达谱 数据和蛋白质的相互作用数据,构建一个全局的异构无向图,通过Katz度量计算lncRNA顶点和蛋白质顶点的相似性,从而依据相似蛋白质的功能标注信息对未知的lncRNA进行功能标注。
如图1所示,本实施例从GENCODE数据库中共下载了15941个lncRNA基因和20284个编码基因。为了获得全基因组范围内的lncRNA和编码基因的联系,分别从COXPRESdb、ArrayExpress等数据库下载了共表达数据,从NPInter数据库下载了lncRNA-蛋白质作用数据。根据这些lncRNA-蛋白质的共表达数据和相互作用数据,采用朴素贝叶斯方法计算lncRNA和蛋白质的相关性:
其中,C(l,p)是基因d(lncRNA)和编码基因p之间的整体相关系数,Cd(l,p)代表l和p在数据集d上的相关分数,D是基因对(l和p)的个数。然后结合计算出的lncRNA和蛋白质的相关性构造lncRNA-蛋白质的关系网络,此网络共包含15941个lncRNA基因和20284个编码基因,并用邻接矩阵LP表示。
从NONCODE2016中下载了lncRNA在人类24个组织中的表达谱数据,根据这些表达谱数据计算lncRNA之间的表达相关性,具体采用皮尔逊相关系数公式计算每对lncRNA之间的表达相关性,然后根据这些表达相关性构造lncRNA相似性网络,此网络共包含15941个lncRNA基因,用邻接矩阵L表示。
根据从STRING数据库下载的蛋白质相互作用数据,构造蛋白质相互作用网络,记作P,共包含20284个蛋白质,结合步骤1、步骤2计算出的矩阵LP、L,构造全局异构网络,用邻接矩阵表示。
Katz度量通过计算两个节点间的距离来衡量两个节点的相似性,基于此,本发明提出通过计算lncRNA节点和蛋白质节点的Katz度量来测量lncRNA基因和蛋白质的相似性,即,利用上述步骤构造的全局网络的邻接矩阵A,计算15941个lncRNA节点和20284个蛋白质节点的Katz度量,计算公式为:
SLP=βLP+β2(L*LP+LP*P)+β3(LP*LPT*LP+L2*LP+L*LP*P+LP*P2) (1)
其中,β是不同长度路径的权重系数,满足β<1/||A||2。计算结果为分数矩阵,分值越大,表示越相似。
对于给定的lncRNA l,从Slp中降序排列的分值中选择前N个蛋白质,并找出前N个蛋白质所对应的注释信息,然后对于每一个GO术语,计算它被指定给lncRNA的概率Pl(Ti),计算公式为:
其中,Slp是lncRNA l和它的邻近编码基因的Kazt相似性分数,Ind(Ti)是一个指示函数,定义如下:
Pl(Ti)越大,则lncRNA l越可能具有该功能。
本发明实施例进行了有效性验证如下。
本发明实施例方法可称之为KATZLGO,需要根据Slp中前N个蛋白质的GO信息来注释RNA,但是,目前没有有效的计算方法确定N的值。在本方法中,通过在手工构建的lncRNA注释数据集lncRNA2GO-55上进行性能评估,根据性能评估的结果选择合适的值,如图2所示。从图2中可以看出,当N取不同的值时,本发明的性能会发生剧烈的波动,当N取值约35至50之间时性能较好,而为40时,性能最好。
本发明实施例比其它预测方法集成了更多生物信息,比如蛋白质相互作用数据。为了评估蛋白质相互作用信息的影响,本发明在825个蛋白质的数据集Protein2GO-825上进行性能评估,如图3所示。显然,含有蛋白质相互作用数据的性能(红色曲线)优于不包含蛋白质相互作用数据的性能(绿线)。
本发明实施例KATZLGO与目前最好的方法LncRNA2Function进行了比较:在数据集lncRNA2GO-55上进行生物过程预测,两种方法的准确率、召回率和F值,如表1所示。在手工注释的55个lncRNA的数据集上,KATZLGO每个性能指标均好于方法LncRNA2Function。
表1
同时,图4示出了在手工标注的55个lncRNA上,本发明实施例和LncRNA2Function分别正确注释的lncRNA的个数比较。图5示出了在全基因组上,本发明实施例和LncRNA2Function分别正确注释的lncRNA的个数比较。图6示出了在不同GO深度下,本发 明实施例和LncRNA2Function分别注释lncRNA的个数比较。
- 还没有人留言评论。精彩留言会获得点赞!