基于相互作用指纹和机器学习的药物靶标的虚拟筛选方法与流程
本发明涉及药物虚拟筛选技术领域。尤其是一种基于相互作用指纹和机器学习的药物靶标的虚拟筛选方法,该方法在传统的分子对接的基础上,通过机器学习对已知活性及非活性小分子与靶标蛋白的相互作用指纹进行训练得出靶标的筛选模型。
背景技术:
在新药发现过程中,虚拟筛选的应用可以提高活性分子的富集,降低筛选的成本。近年来已引起科研机构和制药公司的高度重视。常用的虚拟筛选方法可以分为基于结构的虚拟筛选(SBVS)和基于配体的虚拟筛选(LBVS)。基于配体的虚拟筛选的主要科研精力放在各种分子描述符的产生、相似性的比较。基于配体的虚拟筛选的优势在于速度更快,一般可作为虚拟筛选的最初阶段,劣势在于很难找到不同于已知分子的新的骨架。基于结构的虚拟筛选虽然速度有所下降,但是可以利用靶标的信息,从而有利于全新药物的发现。
分子对接是常用的基于结构的虚拟筛选方法。常用的分子对接软件有:GOLD,FlexX,Glide,Fred,AutoDock,Dock等。分子对接可以分为两步,第一步小分子以不同构象结合到结合口袋,第二步,利用打分函数进行打分。很多研究证明大部分情况下小分子可以找到合适的结合方式,但是打分函数却存在各种问题。按照建立的顺序,常用的打分函数包括:PLP,ChemScore,X-Score,and GlideScore。对接软件中的打分函数,是根据很多已知结合能的蛋白结构利用各种相互作用拟合得到的。
机器学习已经被广泛的应用于药物设计的各个领域,包括靶标预测、毒性预测、药物相似性预测、药物活性预测等。常用的机器学习方法包括支持向量机、决策树、贝叶斯、K邻近和人工神经网络等。
一方面,现有的打分函数很难考虑到不同相互作用之间的耦合作用由于数据集的局限性,另一方面对于特定蛋白来说不具有针对性。最终导致在虚拟筛选中假阳性的概率很高。因此,本领域迫切需要针对特定蛋白的筛选方法,以期提高活性分子的富集程度,提高虚拟筛选的成功率,降低虚拟筛选的成本。
技术实现要素:
本发明的目的在于提供一种基于相互作用指纹和机器学习的药物靶标的虚拟筛选方法,以弥补现有技术的不足。
本发明的目的是这样实现的:
一种基于相互作用指纹和机器学习的药物靶标的虚拟筛选方法,该方法包括以下具体步骤:
步骤1:从CHEMBL、BindingDB或DUD-E数据库或文献中提取靶标的活性数据;
步骤2:对活性分子和非活性分子进行相似性分析,以保证活性与非活性数据的多样性;
步骤3:分子对接,利用薛定谔分子对接软件进行分子对接,每个小分子只保留打分最好的构象;
步骤4:计算找出结合口袋附近的氨基酸残基;
步骤5:计算每个小分子与结合口袋中的氨基酸残基的相互作用能,形成相互作用能矩阵;
步骤6:统计相互作用能矩阵中每个元素出现的概率,去除出现次数较少的元素,形成相互作用指纹;
步骤7:生成支持向量机输入文件,利用网格搜索和交叉验证寻找最优参数;
步骤8:利用步骤7得到的最优参数,交叉验证评估模型;
步骤9:利用步骤7得到的最优参数,训练全部样本,得到筛选模型;
步骤10:利用筛选模型进行虚拟筛选。
所述的步骤1中,提取靶标的活性数据:要求活性小分子的IC50、Kd、Ki值小于10μM,非活性小分子从ZINC数据库中提取。
所述的步骤2中,对活性分子和非活性分子进行相似性分析:要求活性小分子间的ECFP4相似性小于0.8;非活性小分子是以活性小分子为模板挑选的,要求两者物理上相似但化学性质上不相似;以每个活性分子为模板,寻找与活性小分子的分子质量之差不大于20,总的重原子数之差小于2,可旋转键的数目之差不大于1,氢键供体的数目之差不大于1,氢键受体的数目之差不大于2,脂水分配系数之差不大于1的小分子;非活性小分子间的相似性小于0.4,非活性小分子与每个活性小分子的ECFP4相似性小于0.6;最后得到的活性小分子的数目大于100,非活性小分子的数目是活性小分子数目的50倍。
所述的步骤4中,找出结合口袋附近的氨基酸残基:选定靶标PDB自身配体周围范围内的残基作为候选残基,以保证可以包围绝大部分的小分子。
所述的步骤5中,计算每个小分子与结合口袋中的氨基酸残基的相互作用,具体是指计算对接后每个小分子与氨基酸残基的范德华、氢键和疏水相互作用;三种相互作用的定义如下:
1>范德华相互作用为:
其中,i代表结合口袋中的第i个残基,j代表配体小分子中的原子,k代表蛋白质氨基酸残基中的原子,d0代表j原子与k原子的半径之和,djk代表j原子与k原子的实际距离,这里采用了8-4形式的范德华相互作用形式;
2>疏水相互作用为:
其中
其中,i代表结合口袋中的第i个残基,j代表配体小分子中的原子,k代表蛋白质氨基酸残基中的原子,d0代表j原子与k原子的半径之和,djk代表j原子与k原子的实际距离;
3>氢键相互作用为:
要求氢键的供体与受体重原子之间的距离小于供体重原子、供体氢原子、受体重原子三者之间的夹角小于120度;氢键的大小由氢键供体和受体重原子之间的距离确定;
其中,i代表结合口袋中的第i个残基,j代表配体小分子中的原子,k代表蛋白质氨基酸残基中的原子,j与k表示氢键受体或供体中的重原子,djk代表j原子与k原子的实际距离。
所述的步骤5中,形成相互作用矩阵:将结合口袋中氨基酸按照从小到大的方式重新编号,每个氨基酸有范德华、疏水和氢键三种相互作用;形成下表所示的相互作用矩阵。
所述的步骤6中,统计相互作用能矩阵中每个元素出现的频率:如果相互作用矩阵中某一元素出现的频率小于0.1那么去除该维元素。
所述的步骤7中,生成支持向量机输入文件,利用网格搜索和交叉验证寻找最优参数:首先需要将支持向量机软件(libsvm)中的评价指标改为受试者工作特征曲线下面的面积(AUC),其次考虑到数据的不平衡性,在搜索过程中w1参数设定为50,再次,设定SVM训练的核函数为径向基核函数(RBF)。在此基础上利用网格搜索和交叉验证寻找最优参数,需要搜索的参数为(C,γ),设定以下C与γ的参数组合方式:
C=2-5,2-4.5,2-4,......,215;
γ=2-15,2-14.5,2-14,......,25;
其中,C为惩罚因子,γ为RBF核参数;如果多种(C,γ)组合方式都可以使评价指标达到最优,选取C值最小的组合。
所述的步骤8中,利用交叉验证评估模型:利用富集因子和受试者工作特征曲线通过5折的交叉验证对模型进行评估;具体为:
富集因子由以下公式得出
其中,As代表设定的百分位下活性分子的数目,Ds代表选定的百分位下非活性分子的数目,At代表活性分子总体的数目,Dt代表非活性分子的总体数目;公式(4)中,分子计算的是在选定的百分位中活性分子所占的比例,分母计算的是背景分布中活性分子所占的比例。
在实际计算中,分为以下三步:
1>计算理想情况下的富集分布。最理想的情况即全部的活性分子都排在非活性分子之前,按照此分布进行计算。
2>计算分子对接得到的结果。首先对所有对接结果进行排序,然后按照公式(4)进行计算。
3>计算SVM模型预测出来的结果。首先按照预测出来的可能性进行排序,然后按照公式(4)进行计算。
受试者工作特征曲线,根据不同的分界值,以真阳性率为纵坐标,假阳性率为横坐标绘制曲线。其中真阳性率为活性分子被预测为活性分子的比例,假阳性率为非活性分子被预测为活性分子的比例。定义AUC值为受试者工作特征曲线的面积。该值越高表明假阳性的概率越低。
本发明充分利用已知活性和非活性的小分子的相互作用指纹,借助机器学习,构建了一种更加高效的虚拟筛选方法。
本发明的有益效果
相比于传统的方法,本发明的有益效果为:
(1)针对具体靶标进行专项训练,可以充分考虑每种靶标的特异性,避免了传统打分函数拟合不足的缺陷。
(2)计算每个小分子与结合口袋中每个残基的相互作用能,有利于发现有效的结合位点或结合方式。
(3)利用机器学习进行非线性拟合,相较于线性拟合更有利于处理各个相互作用能之间的关联或耦合作用。
(4)结果表明利用本发明,更有利于活性分子的富集。
附图说明
图1为本发明流程图;
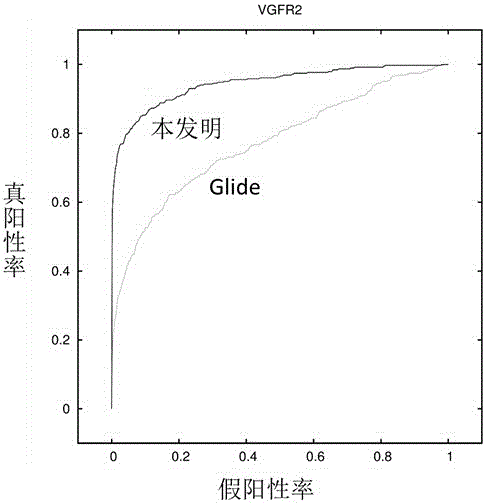
图2为本发明针对靶标VGFR2的Glide与本发明结果的ROC评估图;其中深色实线为本发明对应的受试者工作特征曲线;浅色实线为由Glide分子对接得到的受试者工作特征曲线;
图3为本发明针对靶标VGFR2的Glide与PLEIC-SVM结果的EF评估图;其中虚线为理想的富集曲线,浅色实线为Glide的结果得到的富集曲线,深色实线为利用本发明得到的富集曲线。
具体实施方式
本发明的具体步骤:
(1)从CHEMBL、BindingDB、DUD-E等数据库中提取特定靶标的活性数据。要求活性小分子的IC50、Kd、Ki等活性数据小于10μM,非活性小分子从ZINC数据库中提取。
(2)对活性分子和非活性分子进行相似性分析,以保证活性与非活性数据的多样性。要求活性小分子间的ECFP4相似性小于0.8。非活性小分子是以活性小分子为模板挑选的,要求两者物理上相似但化学性质上不相似。以每个活性分子为模板,寻找与每个活性小分子的分子质量之差不大于20,总的重原子数之差小于2,可旋转键的数目之差不大于1,氢键供体的数目之差不大于1,氢键受体的数目之差不大于2,脂水分配系数之差不大于1的小分子。非活性小分子间的相似性小于0.4,非活性小分子与活性小分子的ECFP4相似性小于0.6。最后得到的活性小分子的数目大于100,非活性小分子的数目是活性小分子数目的50倍。
(3)分子对接。利用薛定谔分子对接软件进行分子对接,每个小分子只保留打分最好的构象。
(4)计算找出结合口袋附近的氨基酸残基。选定靶标PDB自身配体周围范围内的残基作为候选残基,以保证可以包围绝大部分的小分子。
(5)计算每个小分子与结合口袋中的氨基酸残基的相互作用能,形成相互作用能矩阵。计算对接后每个小分子与氨基酸残基的范德华、氢键和疏水相互作用。
(6)统计相互作用能矩阵中每个元素出现的概率,去除出现次数较少的元素,形成相互作用指纹。如果相互作用矩阵中某一元素出现的频率小于0.1那么去除该元素。
(7)生成支持向量机输入文件,利用网格搜索和交叉验证寻找最优参数。
(8)利用(7)中得到的最优参数,交叉验证评估模型。在此利用富集因子和受试者工作特征曲线对模型进行评估。
(9)利用(7)中得到的最优参数,训练全部样本,得到筛选模型。
(10)利用筛选模型进行虚拟筛选。
实施例
结合附图以建立VGFR2靶标的筛选模型为例对本发明进行详细说明。
参阅图1,首先要做的是,更改SVM软件libsvm中的评价指标。从libsvm官方网站下载eval.cpp,eval.h,重新编译,将网格搜索和交叉验证的评估标准改为AUC。
(1)从DUD-E库中收集VGFR2的活性数据,其中包含409个活性小分子,24950个非活性小分子。PDB文件为2P2I。
(2)计算2P2I中自身配体的中心坐标,(38,35,12)。
(3)利用薛定谔分子对接软件Glide进行分子对接。
(4)对接后的每个分子只取GlideScore分数最低的构象。利用glide_ensemble_merge和glide_sort工具实现这一目的。
(5)将分子对接得到的小分子分开,放到文件夹mols里面。
(6)计算找出参考分子以内的氨基酸残基。一共包含60个残基。
(7)对结合口袋的氨基酸残基原子进行归类,氢键供体、氢键受体、疏水原子。计算mols文件夹中每个小分子的原子特征,并计算每个小分子与氨基酸残基的相互作用能,范德华相互作用能、氢键相互作用能、疏水相互作用能。所有的相互作用能信息以每个分子一行的形式追到文件fingers.dat中。
(8)统计不同相互作用能元素出现的频率,删除出现频率较小的元素。生成PLIEIC相互作用能指纹信息,存放到svm.dat中。最终保留了88维信息。
(9)生成支持向量机输入文件,利用libsvm软件工具包中的grid.py工具进行5折的交叉验证和网格搜索。在本实施例中取C等于8,gamma等于1。
(10)利用(9)中得到的最优参数,利用受试者工作特征曲线通过交叉验证评估模型。结果如图2所示,其中黑色的实线表示本发明对应的受试者工作特征曲线,灰色的线表示由Glide分子对接得到的受试者工作特征曲线。由图可以看出,本发明对应的结果得到的受试者工作曲线下的面积(AUC)大于由Glide分子对接得到的AUC值,由此可以得出本发明可以降低假阳性率。
(11)利用(9)中得到的最优参数,利用富集因子通过交叉验证评估模型。首先,根据富集因子的定义,得出理想的EF曲线;然后,对Glide得到的结果进行排序,得到Glide的EF曲线;最后对SVM得出的“可能性”(probability)进行排序,得到本发明对应的EF曲线。结果如图3所示。其中黑色的虚线表示理想的富集曲线,灰色的实线代表Glide的结果得到的富集曲线,黑色的实线代表利用本发明得到的富集曲线。从图中可以看出,利用PLEIC-SVM得到的曲线更接近于理想的富集曲线,即更有利于活性分子的富集。如果以占数据集百分之一的比例为截断,理想结果的富集因子为52,Glide的结果对应的富集因子为22,本发明的结果对应的富集因子为47,结果表明本发明可以使活性分子排在比较靠前的位置,提高活性分子的富集。
- 还没有人留言评论。精彩留言会获得点赞!