一种基于多重进化矩阵的蛋白质二级结构预测方法与流程
本发明涉及生物信息学和传统的蛋白质序列分析技术领域,特别是涉及一种基于多重进化矩阵的蛋白质二级结构预测方法。
背景技术:
蛋白质是生物体内生命活动的主要承担者,是一切生命活动的基础,它的生理功能除了体现在氨基酸构成上还体现在它的空间结构上。因此,预测蛋白质结构是生物信息学领域的一个重要任务。由于蛋白质二级结构是联系蛋白质一级结构和三级结构的纽带,而且也是从一级结构预测其三级结构的关键步骤。当蛋白质二级结构预测正确率达到80%时,就可以准确预测一个蛋白质分子的三维空间结构。可见,蛋白质二级结构预测已经成为研究蛋白质结构和功能的重要手段。
由于已测定结构的蛋白质数量远远小于已知的蛋白质序列数量,并且传统的生物实验测定蛋白质结构的方法费时费力。因此,采用数据驱动的方法(如机器学习技术)来预测未知的蛋白质的结构和功能广受青睐。而影响蛋白质结构类预测效果的关键因素主要集中在两个方面上:一是计算方法,如神经网络、支持向量机(supportvector),深度卷积神经网络技术来进行蛋白质结构预测;二是蛋白质特征信息构造,如正交编码、codon密码子编码法和profile编码法等。
正交编码就是用20位二进制数唯一表示某一种氨基酸,并且满足不同氨基酸的编码向量值的正交积为0,虽然编码方式简单,但是由于其携带生物信息量较少,致使蛋白质二级结构预测准确率较低;codon密码子编码法把氨基酸“还原”成3个碱基的组成形式,而碱基再用二进制数来表示,进而实现结构预测;profile编码是指在氨基酸序列的每个位置上1个氨基酸类型出现的相对概率,能够在一定程度上带有生物进化信息。
目前,现有的传统的蛋白质结构预测方法一般只考虑各种氨基酸在蛋白质序列中所占的比重,存在的不足:这种方法比较简单,但是没有考虑到蛋白质中氨基酸的位置信息和蛋白质进化过程中发生的氨基酸可接受点突变,缺乏对生物进化信息的表示。
综上所述,现有技术中对于预测蛋白质序列中氨基酸残基的二级结构时只考虑氨基酸组成,不能充分考虑蛋白质中氨基酸的位置信息和蛋白质进化过程中发生的氨基酸可接受点突变;分类器参数选择困难及可靠性差等问题,尚缺乏有效的解决方案。
技术实现要素:
为了解决现有技术的不足,本发明提供了一种基于多重进化矩阵的蛋白质二级结构预测方法,其具有能够更为准确的预测蛋白质序列中氨基酸残基的二级结构的效果。
一种基于多重进化矩阵的蛋白质二级结构预测方法,包括以下步骤:
下载蛋白质nr数据库及blast程序本地软件包,生成给定蛋白质序列的位置特异性打分矩阵pssm矩阵,对psi-blast程序进行参数调整得到蛋白质序列的不同趋异度的进化矩阵;
对不同趋异度的进化矩阵对齐特征维度并进行合并,得到多维的特征向量来表示原来的蛋白质序列;
针对得到多维的特征向量,考虑邻近残基的影响,采用滑动窗口方法进行处理得到更多维的特征向量,利用上述多维的特征向量来表示原来的蛋白质,构成多重进化矩阵特征并将多重进化矩阵的元素进行标准化;
将标准化后的多重进化矩阵的特征作为分类器的输入并对分类准确率进行评价,确定相关参数,获得优化模型;
针对结构未知的蛋白质,首先获得该蛋白质的多重进化矩阵特征,进行标准化,将标准化后的多重进化矩阵特征输入优化模型,预测蛋白质的二级结构。
进一步的,psi-blast程序是blast软件包本地化后得到的基于蛋白质序列位置特异性对蛋白质序列进行打分的程序。
进一步的,所述对psi-blast程序进行参数调整得到蛋白质序列的不同趋异度的进化矩阵,在进行参数调整时,根据pam矩阵和blosum矩阵之间的关系,调整psi-blast程序参数,得到不同趋异度的进化矩阵。
进一步的,所述得到不同趋异度的进化矩阵具体过程为:借助nr数据库并且使用psi-blast程序搜索和比对同源序列,psi-blast程序将返回一个20维矢量的pssm,其值是20个氨基酸保守的突变分数,得到的psi-blast的profile是一个lx20的矩阵,也称之为位置特异性得分矩阵(position-specificscorematrix,pssm),其中l是蛋白质序列的长度,在进行参数调整时,根据pam矩阵和blosum矩阵之间的关系,调整psi-blast程序参数,得到不同趋异度的进化矩阵。
进一步的,所述pssm矩阵,其表达公式为:
上述矩阵表示蛋白质进化过程中蛋白质序列第i个位置的氨基酸突变为第j类氨基酸的可能性大小,其值越大表示转成的可能性越大,1≤i≤l,l是蛋白质序列的长度,j从1到20分别表示氨基酸a、r、n、d、c、q、e、g、h、i、l、k、m、f、p、s、t、w、y和v,其中,pssm矩阵中第一列表示原蛋白质序列中氨基酸转换成氨基酸a的可能性得分,第二列表示序列原氨基酸转换成氨基酸r的可能性得分,以此,第三列到第20列分别代表转换成氨基酸n、d、c、q、e、g、h、i、l、k、m、f、p、s、t、w、y和v的可能性;
pssm矩阵第一行表示蛋白质序列第一个位置氨基酸,第二行表示第二个位置上的氨基酸,以此类推,第l行表示第l个位置上的氨基。
进一步的,所述将多重进化矩阵的元素进行标准化时,利用函数把多重进化矩阵的元素标准化到0-1之间。
进一步的,所述确定相关参数时,利用七折交叉验证和网格搜索法优选实验参数,获得优化模型。
进一步的,利用七折交叉验证和网格搜索法优选实验参数,具体步骤为:
(1)设定网格搜索的变量(c,p)的范围以及搜索步距,选择使分类准确率最高的一组c和p;
(2)在寻得了局部最优参数之后,再在这组参数附近选择一个小区间,采用小步距进行二次精搜,再次选择使分类准确率最高的一组c和p;
(3)涉及的所有参数对都用7折交叉验证进行实验,按数据集条数平均分成7份,每次选择其中6份做训练集,剩下的1份做测试集,重复7次。
(4)上述提到的分类准确率的参数对按照以下原则确定:若参数选择过程中有多组c和p对应于最高的验证分类准确率,则选取能够达到最高验证分类准确率中参数c最小的那组c和p作为最佳的参数;如果对应最小的c有多组p,就选取搜索到的第一组c和p作为最佳参数对。
进一步的,所述蛋白质的二级结构进行预测对应的准确率包括整体预测准确率q3及三态预测准确率qi;
其中,整体预测准确率q3指的是被正确预测的3种二级结构的总百分比,其公式如下:
其中:nh,ne和nc分别表示序列中二级结构为h,e和c的残基的总个数,ph,pe和pc分别表示被正确预测为h,e和c构象的残基个数;
三态预测准确率qi来表示每种二级结构被正确预测为h,e或c构象的预测准确率,其计算公式如下式:
其中:pi是待预测序列中被正确预测的处于i构象的残基数目,ni是待预测序列中被正确预测的处于i构象的残基数目,i属于h构象、e构象或c构象。
与现有技术相比,本发明的有益效果是:
1、本发明提出一种新的特征向量构造方法来预测蛋白质二级结构,对于一条蛋白质序列,同时使用多种不同进化趋异度的矩阵来表示蛋白质序列,更为充分的表示了蛋白质结构信息,更全面的考虑了残基替换的可能性,提高了蛋白质二级结构预测的准确率,编码方法简单有效。
2、蛋白质序列特征隐含大量结构和功能信息,对其准确的建模、分析及预测需要强大的算法和计算能力和构造更为完善的特征信息。传统的正交编码、codon编码和profile编码虽然编码简单,但是由于其携带生物信息量较少,致使其二级结构预测的准确率较低,而富含生物进化信息的pssm矩阵具有更大的优势。
3、蛋白质序列残基间存在大量隐式作用影响其二级结构,传统的二级结构预测方法对序列信息的处理能力有限,预测时无法较好的利用序列残基间信息,致使预测结果偏低。由于不同的进化矩阵对不同相关程度的蛋白质序列的敏感性不同。所以多重进化矩阵能够在很好地将序列中氨基酸的位置信息反映出来的同时,考虑序列内部近相关和远相关蛋白质区域之间的相互影响,有效的提高了模型预测的可靠性。
4、实验证明,本发明的方法在大数据样本和使用强分类器时,得到的分类结果更高,提升更明显。
附图说明
构成本申请的一部分的说明书附图用来提供对本申请的进一步理解,本申请的示意性实施例及其说明用于解释本申请,并不构成对本申请的不当限定。
图1为pam矩阵和blosum矩阵概要图;
图2为本方法预测流程图。
具体实施方式
应该指出,以下详细说明都是例示性的,旨在对本申请提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本申请所属技术领域的普通技术人员通常理解的相同含义。
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本申请的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
正如背景技术所介绍的,现有技术中存在对于预测蛋白质序列中氨基酸残基的二级结构时只考虑氨基酸组成,不能充分考虑蛋白质中氨基酸的位置信息和蛋白质进化过程中发生的氨基酸可接受点突变;分类器参数选择困难及可靠性差等的不足,为了解决如上的技术问题,本申请提出了一种基于多重进化矩阵的蛋白质二级结构预测方法。
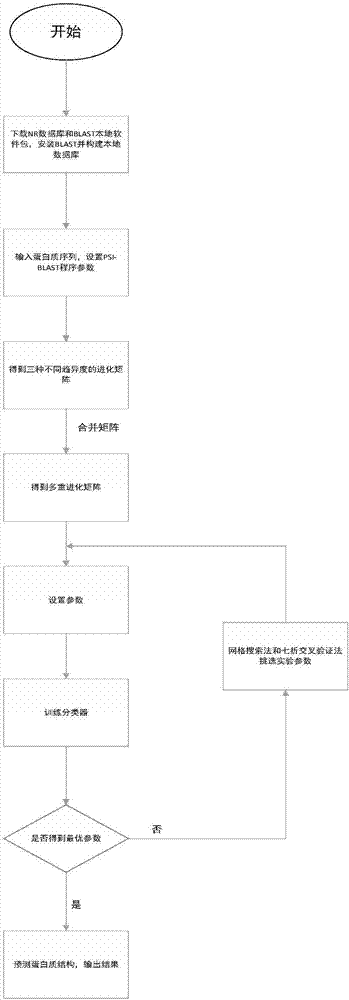
本申请的一种典型的实施方式中,如图1所示,提供了一种基于多重进化矩阵的蛋白质二级结构预测方法,包括如下步骤:
步骤一,其包括:使用psi-blast程序搜索nr数据库生成蛋白质数据集的位置特异性打分矩阵(positionspecificscoringmatrix,pssm),根据pam矩阵和blosum矩阵之间的关系(考虑蛋白质进化过程中发生的氨基酸位点存在突变可能,借助blosum矩阵是基于观测到的远相关蛋白比对得到的打分矩阵,pam矩阵是基于近相关蛋白比对得到的打分矩阵,根据这一特点)调整psi-blast程序参数,得到不同趋异度的进化矩阵;
给定25pdb数据集的一条蛋白质:
>1a1w_
mdpflvllhsvssslssseltelkylclgrvgkrklervqsgldlfsmlleqndlepghtellrellaslrrhdllrrvddfe
要进行计算其位置特异性打分矩阵(pssm矩阵),首先要将blast本地
化:(1)在ncbi上下载blast进行本地配置,本机配置版本:blast-2.3.0+;
(2)在蛋白质数据库ftp://ftp.ncbi.nlm.nih.gov/blast/db/fasta/下载蛋白质数据库nr.gz;
(3)设置psi-blast程序的参数(-num_iterations:3,-eavlue:0.001,-matrix:blosum62)。
通过blast-2.3.0+中的psi-blast程序我们可以得到上述蛋白质的pssm矩阵,其表达公式为:
这个矩阵中第一列表示原蛋白质序列中氨基酸转换成氨基酸a的可能性,第二列表示序列原氨基酸转换成氨基酸r的可能性大小,以此,第三列到第20列分别代表转换成氨基酸n、d、c、q、e、g、h、i、l、k、m、f、p、s、t、w、y和v的可能性。pssm矩阵第一行表示蛋白质序列第一个氨基酸,第二行表示第2个位置上的氨基酸,以此类推。在该参数设置下得到蛋白质序列1a1w_的pssm矩阵第一行为:[-4-5-6-7-5-4-6-6-531-59-3-6-5-4-5-43]。参考了pam矩阵和blosum矩阵之间的关系,如图1所示,通过调整参数-matrix,当设置参数为(-num_iterations:3,-eavlue:0.001,-matrix:blosum90)时,得到蛋白质序列1a1w_的pssm矩阵第一行为:[-5-6-7-8-5-4-6-8-6-20-510-4-7-6-4-6-53]。当设置参数为(-num_iterations:3,-eavlue:0.001,-matrix:pam250)时,得到蛋白质序列1a1w_的pssm矩阵第一行为:[-6-5-6-7-9-6-7-7-711-514-4-7-6-5-9-72]。将这三个20维向量结合,组成一个60维向量来表示蛋白质序列1a1w_的第一个氨基酸,将其余氨基酸的pssm矩阵依次合并。
步骤二,其包括:对步骤一得到的三种不同趋异度的进化矩阵对齐特征维度,组合得到60维的向量来表示原来的蛋白质序列,考虑邻近残基的影响,采用滑动窗口方法对所有特征向量进行了处理,设置滑动窗口为13,得到一个780维向量来表示原来的蛋白质,构成多重进化矩阵特征。
本发明选用蛋白质二级结构字典法(dssp)划分二级结构,并将dssp方法定义的八种结构明确归纳为:h、g属于helices,记作h,e、b属于sheets,记作e,g、s、t、c、i属于coils,记作c。其中:g是310-helix的简称、h是α-helix的简称、i是π-helix的简称、b是isolatedβ-bridge的简称、e是β-stand的简称、s是bend的简称、t是hydrogenbondedturn的简称、rest是apparentlyrandomconformations的简称。
为了能够用计算方法进行训练和预测,需要将相差较大的原始值进行规范化处理。本文利用下面的函数把多重进化矩阵的元素标准化到0-1之间。
其中x是多重进化矩阵中元素的原始值。
步骤三,其包括:将由步骤二得到的规范化后的特征值带入分类器中,利用七折交叉验证对分类准确率进行评价,使用网格搜索法来确定实验参数。实验路线图如图2所示。其具体过程如下:
本方法选择了两个弱分类器(logistics和randomforest)和一个强分类器(m-svmcs)来观察实验结果。现选取强分类器多分类支持向量机m-svmcs来说明实验过程。
在http://www.loria.fr/lauer/msvmpack下载安装最新版本msvmpack软件,里面包含m-svmcs算法。为了对分类器参数进行优化,且保证优化结果的可靠性,本方法结合七折交叉验证与网格搜索法来确定实验参数。
(1)设定网格搜索的变量(c,p)的范围以及搜索步距。其中c的初始范围设置为[2-10,27],p的初始范围设置为[2-10,23]。由于传统方法中步距一般设为0.1,所以改进方法中初始步距选为100倍的步距,设为10。选择使分类准确率最高的一组c和p。
(2)在寻得了局部最优参数之后,再在这组参数附近选择一个小区间,采用传统方法中的小步距进行二次精搜,将步距设为0.1,进行二次参数优化,选择使分类准确率最高的一组c和p。
(3)实验中涉及的所有参数对都用k折交叉验证进行实验,其中k=7。按数据集条数平均分成7份,每次选择其中6份做训练集,剩下的1份做测试集,重复7次。
(4)上述(1)和(3)中提到的分类准确率的参数对按照以下原则确定:若参数选择过程中有多组c和p对应于最高的验证分类准确率,则选取能够达到最高验证分类准确率中参数c最小的那组c和p作为最佳的参数;如果对应最小的c有多组p,就选取搜索到的第一组c和p作为最佳参数对。
步骤四:按照上一步得到的参数设置分类器,输入结构未知的蛋白质序列特征,预测各个位点残基二级结构。
具体的:根据步骤一和步骤二的方法,得到蛋白质的多重进化矩阵特征。使用所获得的优化模型,根据得到的多重进化矩阵特征,预测蛋白质的二级结构。
在预测后,基于已知的蛋白质二级结构数据,采用评估准则评估预测可信度。
关于准确率的整体预测准确率q3:
目前应用最广泛的准确率,它指的是被正确预测的3种二级结构(残基)的总百分比,其公式如下:
其中:nh,ne和nc分别表示序列中二级结构为h,e和c的残基的总个数,ph,pe和pc分别表示被正确预测为h,e和c构象的残基个数。
关于三态预测准确率qi:
我们用qi来表示每种二级结构被正确预测为h,e或c构象的预测准确率。其计算公式如下式:
其中:pi是待预测序列中被正确预测的处于i构象的残基数目,ni是待预测序列中被正确预测的处于i构象的残基数目,i属于h构象、e构象或c构象。
为了使得本领域技术人员能够更加清楚地了解本申请的技术方案,以下将结合具体的实施例与对比例详细说明本申请的技术方案。
实验数据:
(1)实验参数:
经过多组实验,对所获得的实验结果进行对比,选择其中最好的一组作为最优参数。对于数据集cb513和25pdb我们将针对不同方法得到的最优参数汇总如表1所示:
表1.最优参数结果表
(2)实验结果:
关于cb513数据集:
表2.cb513数据集使用blosum62矩阵预测结果
表3.cb513数据集使用多重进化矩阵预测结果
提高的值用粗体标出。通过对比表2和表3可以看出,相比于传统的实验方法,多重进化矩阵这种表示方法在cb513数据集上分别高出了3.65%、0.57%和0.42%。
关于25pdb数据集:
表4.25pdb数据集使用blosum62矩阵预测结果
表5.25pdb数据集使用多重进化矩阵预测结果
通过对比表3和表5可以看出,相比于传统的实验方法,多重进化矩阵这种表示方法在25pdb数据集上分别高出了2.30%、0.54%和1.33%.且通过比对表2——表5中m-svmcs分类器这组实验结果,我们确信本方法在大数据样本和使用强分类器时,得到的分类结果更高,提升更明显。
以上所述仅为本申请的优选实施例而已,并不用于限制本申请,对于本领域的技术人员来说,本申请可以有各种更改和变化。凡在本申请的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本申请的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!