一种大片段插入或缺失的预测方法及系统与流程
本发明涉及生物信息领域,尤其涉及一种应用于靶向测序数据的大片段插入或缺失预测方法及系统。
背景技术:
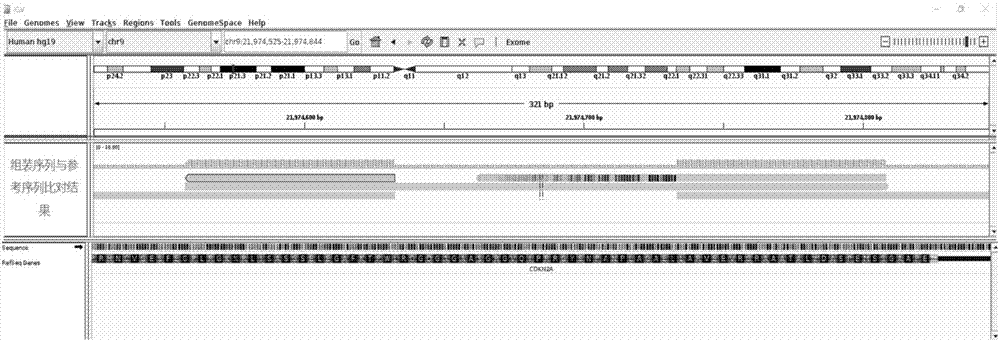
:从2007年第一个个人基因组测序数据诞生之后,测序技术在人类基因组的应用广泛开展。近年来,随着测序技术的不断升级,测序成本也逐年降低,未来即将进入1000元/gb的时代,也使得重测序技术盛行,广泛应用于个人基因组和癌症基因组,用于检测个人遗传疾病基因变异或者癌症基因变异,包括包括点突变、插入和缺失、基因重排等等基因变异。插入和缺失(insertionsanddeletions,indels)是dna和蛋白质在进化过程中发生的序列长度上的改变。当基因序列中缺失在某一段位置的序列时,称为缺失(deletion,del);当基因序列中在某一段位置插入一段序列时,称为插入(insertion,ins)。通常当插入和缺失长度小于10bp时,认为该indels为小片段的插入和缺失,而大于10bp或者更长的片段的插入和缺失,认为indels为大片段的插入和缺失。插入和缺失作为基因变异的一种形式,在疾病的发展中处于非常重要的作用,特别是随着研究的不断深入,大片段的结构插入和缺失作为染色体结构变异的一种表现形式,作用也越来越明显。目前,应用于靶向测序数据的插入和缺失检测方法非常稀少而且准确率不高,大部分方法是应用于全基因组测序数据,而全基因组数据与靶向测序数据有着不同的数据特征,因此这些方法不能直接用于靶向测序数据的插入和缺失的预测,会出现检出结果中假阳性率非常高的情况。而假阳性对后续基因在样本中发挥作用的重要性起着非常关键的作用,因此,亟待出现一种准确度高的应用于靶向测序数据的插入和缺失方法。目前流行的方法是基于序列比对预测indel的方法,即将最原始的测序序列与人类参考基因组进行比对,根据比对结果,来推断插入和缺失的位点信息。近年来,研究者们都在想办法来减少初始比对带来的噪音,但这些尝试始终基于最原始的序列比对结果,很难识别测序序列没有正确错配的区域或者测序序列没有比对到参考基因组的情况,而这些情况正可能是变异发生的区域。另外,第二代测序技术所能检测到的测序序列片段长度在双端75bp~300bp,例如illumina或thermofisher所开发的技术,,受限于该测序序列的长度,目前方法所能正确检测的indel长度比较小,一般为小的插入和缺失,通常能检测的缺失片段长度一般小于30bp,插入片段长度则小于25bp,大片段的插入和缺失基本无法检测。因此,目前基于序列比对预测indel的方法,局限性很大,只能预测片段小的插入和缺失,而无法预测大片段插入和缺失。技术实现要素:针对现有技术中基于靶向测序数据无法准确预测大片段插入和缺失的存在技术缺陷,本发明提供一种可以检测大片段插入和缺失的预测方法,该方法利用组装原理将原始测序序列组装成信息不冗余的组装序列集,该序列集信息准确、长度长、保留了原始序列中的杂合信息,基于该序列的信息来预测大片段插入和缺失,克服了现有技术中只能检测小片段indel的问题,可以检测成百上千片段长度的indel,特别是可以检测插入片段长度大于或等于25bp或缺失片段长度大于或等于50bp,本发明方法运行时间短,预测准确,操作简单。为实现本发明的技术目的,本发明提供一种大片段插入或缺失的预测方法,包括:将待测样本目标区域的多条基因测序序列进行筛选以及碱基错误校正处理,获得待测样本目标区域的多条高质量测序序列;对待测样本目标区域的多条高质量测序序列进行基于冗余信息去除的组装处理,获得保留了原始遗传信息且去冗余信息测序序列的组装序列;将所述组装序列与参考序列进行比对,根据比对结果获得大片段插入或缺失的位置信息;其中,所述待测样本目标区域是利用人类参考基因组中的外显子区域或癌症相关的基因区域得到的。特别是,所述的基于冗余信息去除的组装处理包括:对比所述多条高质量测序序列的首尾碱基信息,得到各个高质量测序序列之间首尾交叠关系;通过对所述各个高质量测序序列之间首尾交叠关系的分析,删除所述各个测序序列之间冗余的首尾交叠关系,得到所述各个高质量测序序列之间简化后的首尾交叠关系;根据所述简化的首尾交叠关系,对所述各个高质量测序序列进行组装,得到所述组装序列,使各个高质量测序序列只有单向的一条近邻序列。其中,所述利用人类参考基因组中的外显子区域或癌症相关的基因区域得到待测样本目标区域包括:对人类参考基因组中的外显子区域或癌症相关的基因区域进行目标筛选,获得包含人类参考基因组中的所有外显子区域或所有癌症相关的基因区域的靶向基因库;根据所述靶向基因库设计探针,将其与待测样本进行杂交,获得目标区域范围内的待测样本dna片段;对所述目标区域范围内的待测样本dna片段进行建库和测序后,得到待测样本目标区域的多条基因测序序列。其中,所述外显子区域是指人类基因组中具有编码功能的dna序列区域,可以是全部人类基因组的外显子区域或人类基因组的部分外显子区域;其中,所述靶向基因区域是指根据需要所选定的基因区域或基因的某些特定的外显子或内含子区域。例如,所述癌症相关的基因区域是指涵盖重要癌变或所有癌变类型相关的基因区域。癌症相关的基因是指现有技术中由于一个或多个基因发生序列、结构、或其表达和/活性发生变化导致其与正常基因相比有缺陷的基因,致使细胞癌变。其中,所述根据所述靶向基因库设计探针是基于靶向基因库中的基因而设计的,采用常规设计方法进行,设计的探针应该满足特异性的要求,从而实现获取目标区域范围内的基因的目的。其中,所述将其与待测样本进行杂交,获得目标区域范围内的待测样本dna片段的方法采用生物学方法,具体的杂交过程依据生物学领域常用的方法进行,例如根据《分子生物学》操作。其中,所述经建库和测序步骤,得到待测样本基因序列是根据dna建库试剂盒的说明书进行。特别是,在将待测样本的dna片段进行建库及测序之前,先进行dna片段化处理,该步骤可以使用片段化仪器进行,例如covarism220。其中,所述将待测样本目标区域的多条基因测序序列进行筛选包括:去掉含有接头序列的所述测序后的基因序列;根据设置的碱基质量阈值,去除低于碱基质量阈值的测序,得到碱基质量均大于阈值的多条基因测序序列。其中,所述碱基质量阈值可以是20或30。其中,所述去除低于碱基质量阈值的测序是指,如果一条测序序列有一个或多个碱基质量值低于20,则过滤该序列,得到碱基质量均大于20的高质量待测样本序列,同理,碱基质量阈值还可以是30。当碱基质量值为20时,认为该碱基的准确率为99%。还可以调整该碱基质量阈值为30,即保留准确率大于99.9%的测序序列为高质量序列。本发明的碱基质量筛选适用于较高的测序深度,比如测序深度大于500x~1000x以上的序列,可以降低系统测序错误造成的影响。其中,所述将将待测样本目标区域的多条基因测序序列进行碱基错误校正处理包括:收集碱基质量均大于阈值的待测样本序列中所有高频k-mer;对每一条测序序列进行修正使序列上每一个k-mer均为高频。特别是,所述大片段是长度≥10bp的基因片段。特别是,所述大片段是长度≥25bp的基因片段。特别是,所述大片段是长度≥50bp的基因片段。特别是,所述大片段是长度≥100bp的基因片段。其中,所述大片段为插入片段或缺失片段。其中,本发明所称人类参考基因组序列可以从公共数据库进行下载,如ucscgenomebrowser或ncbigenomeresources,目前常用的版本为hg19或hg38。为实现本发明的技术目的,本发明再提供一种预测大片段插入或缺失的系统,包括:筛选模块,用于筛选待测样本目标区域的多条基因测序序列,获得筛选后的多条基因测序序列;碱基校正模块,用于将从筛选模块筛选获得的多条测序序列进行碱基错误校正的,获得碱基校正后的多个高质量测序序列;组装模块,用于将从碱基校正模块获得的多个高质量测序序列进行基于冗余信息去除的组装处理,获得保留了原始遗传信息且去冗余信息测序序列的组装序列;以及分析模块,用于将组装模块获得的保留了原始遗传信息且去冗余信息测序序列的组装序列与参考序列进行比对的,获得大片段插入或缺失的位置信息。其中,所述组装模块包括:比对单元,用于对比所述多条高质量测序序列的首尾碱基信息,得到各个高质量测序序列之间首尾交叠关系;判断单元,用于分析比对单元获得的各个高质量测序序列之间首尾交叠关系,删除所述各个测序序列之间冗余的首尾交叠关系,得到所述各个高质量测序序列之间简化后的首尾交叠关系;连接单元,用于根据判断单元得到的简化后的首尾交叠关系,对所述各个高质量测序序列进行组装,得到所述组装序列,使各个高质量测序序列只有单向的一条近邻序列。为实现本发明的目的,本发明还提供一种体现在计算机可读介质中的计算机程序产品,当在计算机上执行时,执行步骤包括:将待测样本目标区域的多条基因测序序列进行筛选以及碱基错误校正处理,获得待测样本目标区域的多条高质量测序序列;对待测样本目标区域的多条高质量测序序列进行基于冗余信息去除的组装处理,获得保留了原始遗传信息且去冗余信息测序序列的组装序列;将所述组装序列与参考序列进行比对,根据比对结果获得大片段插入或缺失的位置信息;其中,所述待测样本目标区域是利用人类参考基因组中的外显子区域或癌症相关的基因区域得到的。特别是,所述的基于冗余信息去除的组装处理包括以下执行操作:对比所述多条高质量测序序列的首尾碱基信息,得到各个高质量测序序列之间首尾交叠关系;通过对所述各个高质量测序序列之间首尾交叠关系的分析,删除所述各个测序序列之间冗余的首尾交叠关系,得到所述各个高质量测序序列之间简化后的首尾交叠关系;根据所述简化的首尾交叠关系,对所述各个高质量测序序列进行组装,得到所述组装序列,使各个高质量测序序列只有单向的一条近邻序列。其中,所述利用人类参考基因组中的外显子区域或癌症相关的基因区域得到待测样本目标区域包括:对人类参考基因组中的外显子区域或癌症相关的基因区域进行目标筛选,获得包含人类参考基因组中的所有外显子区域或所有癌症相关的基因区域的靶向基因库;根据所述靶向基因库设计探针,将其与待测样本进行杂交,获得目标区域范围内的待测样本dna片段;对所述目标区域范围内的待测样本dna片段进行建库和测序后,得到待测样本目标区域的多条基因测序序列。其中,所述外显子区域是指人类基因组中具有编码功能的dna序列区域,可以是全部人类基因组的外显子区域或人类基因组的部分外显子区域;其中,所述靶向基因区域是指根据需要所选定的基因区域或基因的某些特定的外显子或内含子区域。例如,所述癌症相关的基因区域是指涵盖重要癌变或所有癌变类型相关的基因区域。癌症相关的基因是指现有技术中由于一个或多个基因发生序列、结构、或其表达和/活性发生变化导致其与正常基因相比有缺陷的基因,致使细胞癌变。其中,所述根据所述靶向基因库设计探针是基于靶向基因库中的基因而设计的,采用常规设计方法进行,设计的探针应该满足特异性的要求,从而实现获取目标区域范围内的基因的目的。其中,所述将其与待测样本进行杂交,获得目标区域范围内的待测样本dna片段的方法采用生物学方法,具体的杂交过程依据生物学领域常用的方法进行,例如根据《分子生物学》操作。其中,所述经建库和测序步骤,得到待测样本基因序列是根据dna建库试剂盒的说明书进行。特别是,在将待测样本的dna片段进行建库及测序之前,先进行dna片段化处理,该步骤可以使用片段化仪器进行,例如covarism220。其中,所述将待测样本目标区域的多条基因测序序列进行筛选包括以下执行步骤:去掉含有接头序列的所述测序后的基因序列;根据设置的碱基质量阈值,去除低于碱基质量阈值的测序,得到碱基质量均大于阈值的多条基因测序序列。其中,所述碱基质量阈值可以是20或30。其中,所述去除低于碱基质量阈值的测序的执行步骤是,如果一条测序序列有一个或多个碱基质量值低于20,则过滤该序列,得到碱基质量均大于20的高质量待测样本序列,同理,碱基质量阈值还可以是30。当碱基质量值为20时,认为该碱基的准确率为99%。还可以调整该碱基质量阈值为30,即保留准确率大于99.9%的测序序列为高质量序列。本发明的碱基质量筛选适用于较高的测序深度,比如测序深度大于500x~1000x以上的序列,可以降低系统测序错误造成的影响。其中,所述将将待测样本目标区域的多条基因测序序列进行碱基错误校正处理包括以下执行步骤:收集碱基质量均大于阈值的待测样本序列中所有高频k-mer;对每一条测序序列进行修正使序列上每一个k-mer均为高频。特别是,所述大片段是长度≥10bp的基因片段。特别是,所述大片段是长度≥25bp的基因片段。特别是,所述大片段是长度≥50bp的基因片段。其中,本发明所称人类参考基因组序列可以从公共数据库进行下载,如ucscgenomebrowser或ncbigenomeresources,目前常用的版本为hg19或hg38。本发明的优点:1、本发明公开的预测大片段插入和缺失的方法,应用于靶向基因测序数据,运行时间更短,预测结果更准确,一旦检出,结果肯定为真,准确率为99.99%,假阳性率极低。2、本发明公开的方法,充分考虑了测序序列之间的关联性,充分利用了最原始的测序序列信息,保留了原始序列中的杂合信息,在组装时将测序序列间相互比对,避免与参考序列差异造成的错误,因此本发明方法可以得到较长的无测序错误拼接序列,从而准确预测大片段indel。附图说明:图1是本发明实施例1的组装处理流程图,图中,a表示交叠图,b表示简化关系图,c表示最终得到的组装序列;图2是本发明试验例1的组装序列与参考序列比对结果图;图3是本发明试验例1的获得的测序序列的琼脂凝胶电泳图。具体实施方式下面结合具体实施例,进一步阐述本发明。但这些实施例仅限于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体实验条件的实验方法,通常按照常规条件,或按照厂商所建议的条件。在下面的描述中阐述了很多具体的细节以便于充分理解本发明,但是,本发明还可以采用其他不同于在此描述的其他方式来实施,因此,本发明并不限于下面公开的具体实施例的限制。实施例1待测样本的基因序列的获得1、靶向基因库的获得1.1样本处理以研究对象或检测对象得到待测样本,进行dna提取,利用定量仪器(比如nanodrop仪器)进行定量,从而确定待测样本dna质量是否达到后续的测序要求。利用试剂盒进行样本dna提取,继而进行dna扩增,分别得到足量的待测样本dna。其中,待测样本可以是来源于动物、人类,特别是病人的正常样本或肿瘤样本,样本类型包括但不限于ffpe(石蜡包埋的样本)、组织(从病人身体上切割的组织)样本、血液(从病人身上抽取的血液)样本中的一种,根据样本类型不同,选择不同的试剂盒待测样本dna的提取,例如ffpe样本用dnaffpe试剂盒进行提取,血液样本用dnablood试剂盒进行提取。1.2靶向测序筛选研究或检测关注的靶向区域,可以是全部基因的外显子区域或某些基因外显子的某一些区域或主导/介导了癌症发生、发展等过程的、起决定性作用的癌症基因,组成靶向基因库,根据靶向基因库中的基因设计探针,利用杂交原理将设计的探针分别与步骤1.1得到的待测dna样本进行杂交,筛选获得待测样本目的基因片段。根据测序仪测序的操作手册,对获得待测样本目的基因片段进行建库操作,即,将杂交获得待测样本目的基因片段,利用片段化仪器(例如covarism220)进行dna片段化,并使用dna建库试剂盒进行建库操作,通过磁珠纯化使dna片段长度更集中,最后对纯化后的dna片段进行pcr扩增,获得足够浓度和质量的dna,得到待测样本dna文库,对待测样本dna文库进行测序,测序过程中先进行文库质量检测,例如选用dsdnahsassay试剂盒进行,检测质量合格之后,再利用测序仪(例如,illumina服务商提供的测序仪miseq/nextseq等或thermofisher服务商提供的ionproton等)对文库进行基因测序,得到待测样本目的基因序列。其中,所述筛选主导或介导了癌症发生、发展等过程的、起决定性作用的基因包括但不限于,egfr基因(epidermalgrowthfactorreceptor,简称为egfr,表皮生长因子受体基因)、alk(anaplasticlymphomakinase,简称alk,间变性淋巴瘤激酶)、braf(serine/threonine-proteinkinase,简称braf,丝/苏氨酸特异性激酶)等。实施例2大片段插入和缺失的预测大片段插入和缺失,即发生了大片段长度的dna序列插入和缺失现象,优选地,插入长度≥25bp和缺失片段长度≥50bp。算法操作流程如下:1、选择高质量测序序列将实施例1获得的待测样本目的基因序列进行数据筛选,筛选方法如下:1)去除含有接头序列的测序序列;如果测序序列中含有接头序列,例如接头序列为agatcggaagagcacacgtc,测序序列为agatcggaagagcacacgtcaacgctaggcatcgat,则去掉该条测序序列。2)保留高质量的测序序列;如果一条测序序列有一个或多个碱基质量值低于20,则过滤该序列,得到碱基质量均大于20的高质量待测样本序列。碱基质量值为20时,认为该碱基的准确率为99%。根据需要可以调整该碱基质量阈值。例如,选择碱基质量值为30,即保留准确率大于99.9%的测序序列为高质量序列。本发明的碱基质量筛选适用于较高的测序深度,比如测序深度大于500x~1000x以上的序列,可以降低系统测序错误造成的影响。2、校正碱基错误收集碱基质量均大于20的待测样本序列中所有高频k-mer,然后对每一条测序序列进行修正使序列上每一个k-mer均为高频的。同样地,根据需要可以调整该碱基质量阈值,例如,将阈值设定为30。3、序列组装该步骤如图1所示,包括:s101对比所述多条高质量测序序列的首尾碱基信息,得到各个高质量测序序列之间首尾交叠关系;s102通过对所述各个高质量测序序列之间首尾交叠关系的分析,删除所述各个测序序列之间冗余的首尾交叠关系,得到所述各个高质量测序序列之间简化后的首尾交叠关系;s103根据所述简化的首尾交叠关系,对所述各个高质量测序序列进行组装,得到所述组装序列,使各个高质量测序序列只有单向的一条近邻序列。传统的方法将一条序列打断成固定长度为k的多条序列(即k-mer原理),再寻找这些固定长度的多条序列之间有重叠,根据重叠情况构建简化图,这种方法需要很大的计算量,一般需要的计算复杂度为o(n2),n为所有测序序列长度的总和。而本发明方法采用双向fm-index这一用于全文索引的数据结构来加速这一过程,去掉了冗余的重叠,极大的减少了内存要求,使得计算复杂度降低到了o(n)。因此,算法的应用使得整个方法及系统的运行速度大大加快,减少了对硬件设备的硬性要求配置。例如,处理370mb的序列在14个内核,内存为512mb的硬件配置上的时间为25.4秒,处理速度非常快。这一速度的提升,大大缩短了病人变异信息的获得时间,从而使得病人在最快的速度进行针对性地用药,减少疾病窗口期。如图1所示,1、2、3、4、5、6、7、8、9、10示例为依次测得的每一条测序序列,根据测序仪产生的数据,可以是一条75bp长度的测序序列或者长度为150bp的测序序列,其中第6条和第8条序列带有相同变异信息,第5条和第7条序列带有相同的变异信息。首先通过对比所述多条序列的首尾碱基信息确定多条序列的首尾交叠关系,构建得到交叠图1中a,即根据序列碱基之间的重叠情况判定各条测序序列之间的交叠情况,图上的各点之间存在直接或间接推导关系,实线为可保留的关系,虚线为冗余的关系信息,所述冗余的关系信息可以通过实线间的关系进行推导获得;其次,根据关系信息,去掉冗余关系信息,从而得到简化关系图1中b;最后,将点与点之间的序列进行连接,获得更长的组装序列,最后得到如图1中c所示的仅有4条序列。而从以上组装关系图中可以看出,最原始测序序列中存在的第6条和第8条序列的变异信息以及第5条和第7条序列被合并为两条携带有变异信息的组装序列,因此可见,最终的组装序列保留了最初原始测序序列所携带的变异信息,最大程度的保留了这部分信息,去除冗余信息,从而,得到准确的组装序列。为后续的大片段插入和缺失提供了准确预测的基础。4、大片段indel的位置预测将组装序列与人类参考序列进行序列比对,基于比对结果,通过检测断点信息输出判断待测样本发生indel的位置信息,包括染色体、indel起始位置、indel终止位置以及indel长度等信息。其中,插入长度和缺失长度>=10bp可以称为大片段缺失。优选地,插入片段长度≥25bp或缺失片段长度≥50bp。其中,人类参考基因组序列可以从公共数据库进行下载,如ucscgenomebrowser或ncbigenomeresources,目前常用的版本为hg19或hg38。试验例1实际病人样本大片段插入和缺失分析取病人a的肿瘤样本进行dna提取,提取方法采用常规技术进行,获得足量的肿瘤样本dna。取适量肿瘤dna样本,利用本发明实施例1中提到的方法进行建库测序,将得到的测序序列分别用实施例1中提到的om-indel算法和常用的indel预测软件pindel(下载地址:http://gmt.genome.wustl.edu/packages/pindel/)进行indel变异分析。将预测出来的indel根据区域设计探针进行pcr,将扩增产物采用一代测序方法进行测序,测序结果如图3所示,预测结果如表1及图2所示:表1病人a样本indel变异分析结果indel位置变异类型基因indel长度(bp)实施例1pindel一代测序chr9:21974630-21974733delcdkn2a1041未检出存在其中:表1中del代表区域发生缺失现象表中的数据“1”表示存在染色体变异。从图2(igv软件查看)组装序列与参考基因组序列比对结果可以看出,共有5条组装序列在indel区域,从这5条的情况可以推断出在chr9:21974630-21974733的位置确实存在缺失。表2组装序列的比对情况详细组装序列cigar(比对的情况)unitig171s75munitig2251munitig374m72hunitig4414munitig5382m根据图3的胶图可以看到,正常应该产生的pcr产物片段长度为483,而除此产物外,还存在片段长度为381的pcr产物,因此,可以推断存在片段长度为102bp的缺失,断而利用一代测序仪进行测序后,进行峰图序列的分析,判断在chr9:21974630-21974733存在102bp的缺失。该结果与表1测定结果即本发明方法预测结果一致,一代测序方法检测indel是目前最为可靠的验证方法,可以认为该病人确实存在以上长indel情况,而pindel算法无法预测出这些大片段indel的存在。而缺失的存在具有非常重要的意义,该缺失为基因cdkn2a基因的缺失,该基因为细胞周期依赖性激酶抑制基因,是在肿瘤中非常重要的抑癌基因,与多种癌症的发生相关,根据预测出来该基因的大片段缺失,便于推断疾病发生的原因及针对性地进行用药。另外,本方法样本总共产生的测序序列数据为400mb,利用本实施例1提供的方法单cpu的运行时间为748.86秒,而pindel的运行时间为8557.375秒,为实施例1中方法的将近十倍,随着数据量的不断增大,pindel的缺陷将更加明显,因此本发明提供的方法从速度上有明显的优势。试验例2,已知大片段的插入和缺失ccrf-cem细胞系中存在已知的insertion,即第9条染色体上139399385位置存在插入片段且片段长度为36bp。将该细胞系样本进行dna提取,提取方法采用常规技术进行,获得足量的细胞系样本dna。取适量细胞系样本dna样本,利用本发明实施例1中提到的方法进行建库测序,将得到的测序序列分别用实施例1中提到的om-indel算法和常用的indel预测软件pindel(下载地址:http://gmt.genome.wustl.edu/packages/pindel/)进行indel变异分析,预测结果如下:表3ccrf-cem细胞系样本indel变异分析结果可见,本发明方法获得的结果与已知的indel信息一致,证明该方法能对大片段的插入和缺失进行检测。试验例3公开大数据大片段插入和缺失的检测我们采用千人基因组中na12878外显子区域的测序数据利用实施例1中提到的om-indel算法和常用的indel预测软件pindel(下载地址:http://gmt.genome.wustl.edu/packages/pindel/)进行indel变异分析。将预测出来的indel根据区域设计探针进行pcr,将扩增产物采用一代测序方法进行测序,预测结果如下:表4公开数据indel变异分析结果其中:表4中del代表区域发生缺失现象,ins代表区域发生插入现象。表中的数据“1”表示存在染色体变异。由表4可以看出,本方法总共预测出30个在外显子区域的大片段插入和缺失,包括22个缺失失和8个插入,片段长度非常长,缺失片段长度都大于30bp且最长的缺失片段长度达到了6684bp,插入片段长度都大于25bp且最大的插入片段长度达到了96bp。而且,本发明方法获得的结果与一代测序结果一致,证明该方法能对大片段的插入和缺失进行检测。从以上例子可以看出,本发明方法获得的结果与一代测序结果一致,而且比一代测序更加便捷,通量更大,并且可以检测未知的indel。与常用的indel算法pindel相比较,本发明所提供的方法可以预测更长的indel,包括缺失长度≥50bp或者插入长度≥25bp的片段,而pindel具有明显的算法缺陷,不能预测大片段的indel。因此,本发明方法预测的结果,可以作为正确的indel结果,进行基因解读。目前流行的indel方法是将原始的测序序列比对到参考序列,然后检测两个序列之间的差别,这种方法我们称之为基于比对的变异检测(如pindel)。它操作相对简单,利用比对结果来识别可能的indel。但是,在差异较大的区域,短测序序列造成的错误比对往往会误导变异检测,从而导致无法预测更大片段的indel或者预测出来的indel具有明显错误。而本发明的方法改变了以往的预测方法,开创性了将组装概念应用于针对靶向测序数据来预测大片段的插入和缺失方法。具体地,本申请在对序列进行组装时,在测序序列间相互比对,避免与参考序列差异造成的错误;从而得到组装后的较长的无测序错误拼接的序列,将这样的长序列比对到参考序列可以极大降低比对错误和由其引起的变异检测错误。与以往的indel方法相比,本发明的组装的算法有明显优势,尤其在长indel方法上,这是因为长indel更容易造成短序列的比对错误。而且,预测结果准确,经过多种数据验证,本方法得到的结果正确无误且无假阳性结果。以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之类。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1