用于有机分子晶体结构预测中高精度能量排位方法与流程
本发明属于有机分子晶体结构技术领域,具体涉及用于有机分子晶体结构预测中高精度能量排位方法。
背景技术
化合物具有形成不同的晶体结构的特点被称为多晶型现象。而化合物本身的关键理化性质如密度,形态,溶解度,溶解速率等受到其晶型的强烈影响。对于药品,晶型可以强烈影响药物的生物利用度,最终影响药物的治疗表现。实验多晶型药物筛选已成为标准药物研发过程中一个必不可少的环节。实验中,人们手动或借助机器人手动设置关键结晶参数,但正确的结晶条件很难通过实验在短时间内获得。一种可选择的方案是通过计算机模拟对药物分子进行晶体结构预测(csp),找到潜在的多种稳定晶型,再针对少数目标明确的潜在晶型进行实验。
在过去的十年中,无机和有机晶体预测(csp)都取得了巨大的进步。尽管有许多相似之处,但无机和有机晶体预测需要面对截然不同的挑战。无机csp中,人们关心的是键合和电子性质,而有机csp更关心结构改变和相变。药物研发则与有机分子的csp有关。这个领域目前有两个主要挑战,一个是对晶体空间采样的完备性,另一个是晶体结构能量排位的准确性。第一个挑战已经有了很大的进步,但第二点,结构的精确能量排位仍然是制约csp在药物工业中大规模应用的瓶颈。
准确计算不同低能晶体结构间的微小能量差需要高精度的量子力学计算,而高精度量子力学计算的时间复杂度随体系电子数n的增长是o(n3)~o(n4),当非对称单元内独立分子个数增多,且空间群操作数增多时,对csp过程中产生的大量晶体结构进行高精度能量计算就成为了csp的瓶颈。
技术实现要素:
针对上述技术问题,本发明提供一种对有机分子晶体结构预测过程中产生的大量晶体结构进行快速精确能量计算的方法,以提高晶体结构能量排位的效率和精度。为了达到以上目的,使用孤立大分子团簇体系能量计算中所使用的密度分块相互作用算法和周期性体系的qm/mm相结合的方法,设计了适用于周期性有机分子晶体体系的高效精确能量计算方法。该方法计算中所设计的框架,可适用于各种量子力学精度的第一性原理计算方法,泛函和基组。
所采用的技术方案为:
用于有机分子晶体结构预测中高精度能量排位方法,包括以下步骤:
(1)确定中心单胞的量子力学作用半径
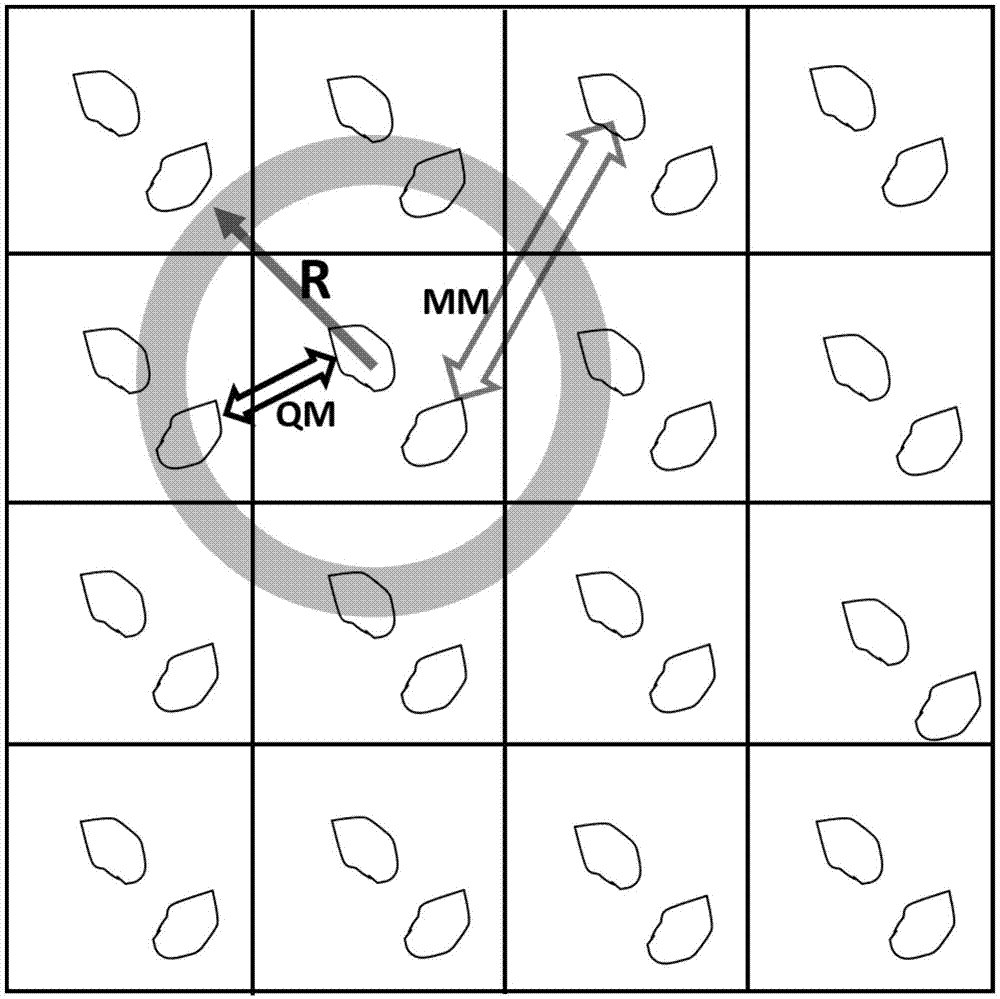
计算中心单胞中的分子内所有原子与周围扩展单胞分子中原子的范德华半径和,以此半径和加1.5埃作为截断,找到距离中心单胞最近的一圈分子,以中心单胞的几何中心到这一圈分子中最远的距离作为量子力学作用半径r;
(2)中心单胞内,采用密度分块相互作用算法进行能量计算
在量子力学精度下,分别计算各个分子的能量,此时包含其余分子产生的静电势作用,静电势由其余各分子的原子核势和电子密度分布产生,静电势和各个分子的电子密度分布通过彼此迭代直到收敛获得;迭代收敛后的能量为:
中心单胞内的能量eiso[ρ],其中ρ代表电子密度,这个能量由三部分构成:第一项是每个孤立分子的能量求和,其中
(3)半径r的范围内,中心单胞外的分子对中心单胞内分子的作用能在量子力学精度下计算,仍采用第2步的迭代求解的流程,但只计算这部分分子对中心单胞分子的作用能:
分子对中心单胞分子的作用能einter_ij_qm包括:第一项是原子核之间的静电相互作用,第二项是电子之间的相互作用;
(4)在经典分子力学精度条件下,计算半径r范围之外,周期性扩展单胞对中心单胞内分子的作用能;这部分能量由周期性扩展晶胞中的分子在中心单胞处产生的长程静电势对中心单胞中总的电子密度积分得到,其计算公式为:
其中
(5)计算晶体总能量,晶体总能量包括中心单胞内的能量,以及中心单胞与周期性扩展单胞之间的能量:
e=eiso+eperiodic
其中,中心单胞的能量采用第(2)步计算的能量,而周期性扩展单胞对中心单胞的作用能包括第(3)步和第(4)步计算的能量和:
其中,eitner_ij_qm即第(3)步中所计算能量,求和条件
本发明提供的用于有机分子晶体结构预测中高精度能量排位方法,具有的技术效果有:
(1)提高了药物分子晶体中能量计算效率,将晶体结构量子力学精度级的计算时间复杂度从o(n3)~o(n4)降到o(n);
(2)保障了晶体搜索方向的正确性,精准的能量反馈,在晶体预测过程,将引导csp在正确的方向上寻找真正的低能优势晶型。
附图说明
图1是实施例确定量子力学作用半径示意图;
图2是实施例的密度分块相互作用算法流程图。
具体实施方式
结合实施例说明本发明的具体技术方案。
用于有机分子晶体结构预测中高精度能量排位方法,包括以下步骤:
(1)确定中心单胞的量子力学作用半径
计算中心单胞中的分子内所有原子与周围扩展单胞分子中原子的范德华半径和,以此半径和加1.5埃作为截断,找到距离中心单胞最近的一圈分子,以中心单胞的几何中心到这一圈分子中最远的距离作为量子力学作用半径r;如图1所示;
(2)中心单胞内,采用密度分块相互作用算法进行能量计算
在量子力学精度下,分别计算各个分子的能量,此时包含其余分子产生的静电势作用,静电势由其余各分子的原子核势和电子密度分布产生,静电势和各个分子的电子密度分布通过彼此迭代直到收敛获得;如图2所示;迭代收敛后的能量为:
中心单胞内的能量eiso[ρ],其中ρ代表电子密度,这个能量由三部分构成:第一项是每个孤立分子的能量求和,其中
(3)半径r的范围内,中心单胞外的分子对中心单胞内分子的作用能在量子力学精度下计算,如图1所示,仍采用第2步的迭代求解的流程,但只计算这部分分子对中心单胞分子的作用能:
其中分子对中心单胞分子的作用能einter_ij_qm包括:第一项是原子核之间的静电相互作用,第二项是电子之间的相互作用;
(4)在经典分子力学精度条件下,计算半径r范围之外,周期性扩展单胞对中心单胞内分子的作用能;这部分能量由周期性扩展晶胞中的分子在中心单胞处产生的长程静电势对中心单胞中总的电子密度积分得到,其计算公式为:
其中
(5)计算晶体总能量,晶体总能量包括中心单胞内的能量,以及中心单胞与周期性扩展单胞之间的能量:
e=eiso+eperiodic
其中,中心单胞的能量采用第(2)步计算的能量,而周期性扩展单胞对中心单胞的作用能包括第(3)步和第(4)步计算的能量和:
其中,eitner_ij_qm即第(3)步中所计算能量,求和条件
- 还没有人留言评论。精彩留言会获得点赞!