一种K+响应型两亲嵌段共聚物载药胶束及其制备方法与流程
本发明属于刺激响应型胶束领域,特别涉及一种新的K+响应型两亲嵌段共聚物载药胶束及其制备方法。
背景技术:
嵌段共聚物胶束是近年来快速发展的一种新型药物纳米载体。在水溶液中,两亲嵌段共聚物中的疏水嵌段构成内核,亲水嵌段构成外壳,形成具有球形内核-外壳结构的嵌段共聚物胶束。嵌段共聚物胶束作为药物载体具有以下优势:(1)临界胶束浓度(CMC)较低,具有高度的热力学和动力学稳定性,即使在CMC以下也不易破坏,具有很高的耐稀释能力;(2) 在水溶液中自组装形成胶束,内核可以装载疏水性的药物,亲水性的表面使其不易被网状内皮系统(RES)识别和获取,延长药物在血液中的循环时间;(3)尺寸小,分布窄,一般在 10~200nm之间,可避免被肾脏清除;(4)可通过EPR效应穿透肿瘤部位的毛细血管壁进入肿瘤组织,在肿瘤部位蓄积,从而达到靶向释药,减轻药物的毒副作用。
随着高分子材料科学和纳米技术的发展,越来越多的嵌段共聚物胶束被用作药物载体,有部分药物载体已进入临床试验阶段乃至被批准临床使用。为最大程度地提高治疗可能性并降低药物的毒副作用,通过对聚合物结构的灵活设计,实现了多种不同刺激响应型嵌段共聚物胶束结构的构建,如温度响应型、pH响应型、光响应型、磁场响应型、超声响应型、化学与生物活性分子响应型等。然而,pH、光、磁场、超声等常见刺激响应型胶束存在药物利用率低、释药行为控制有限等问题,化学与生物活性分子响应型胶束应用较为局限,靶向控释效果不佳。
基于此,申请号为201510299844.5的专利申请中提出了“K+响应型靶向胞内释药的两亲嵌段共聚物载药胶束及其制备方法”,将苯并15冠5丙烯酰胺引入嵌段高分子,设计并制备了一种 K+响应型靶向胞内释药的两亲嵌段共聚物载药胶束。但是,这种K+响应型嵌段共聚物胶束仍存在一定的不足和缺陷:一是嵌段共聚物本身结构构建较为复杂,合成步骤较多,需引入丙烯酰胺这种亲水物质以调节整个嵌段共聚物的低临界溶解温度(LSCT),且调节过程比较繁琐;二是载药胶束中嵌段共聚物在胞外的的低临界溶解温度(LCST)在生理温度37℃之上,胶束保持原有的形态,而进入细胞内响应K+后,嵌段共聚物的LCST向低温迁移,在生理温度37℃之下,此时嵌段共聚物的亲水端由溶胀状态变为收缩状态,胶束结构解离,使得整个嵌段共聚物收缩为紧密的胶粒状结构,这种胶粒结构在体内团聚后粒径变大,难以被肾脏清除,在体内蓄积可能引发潜在的毒性;三是载药胶束响应K+后,整个嵌段共聚物收缩为紧密疏水的胶粒状,这种团聚的胶粒结构会包裹一定的疏水药物,降低药物利用率。
技术实现要素:
本发明的目的在于克服现有K+响应型载药胶束的不足,提供一种新的K+响应型两亲嵌段共聚物载药胶束及其制备方法,所述载药胶束在体内释药后其两亲嵌段共聚物可经正常代谢途径排出体外,以避免在体内蓄积引发潜在毒性,并能提高药物利用率,简化合成方法。
本发明所述K+响应型两亲嵌段共聚物载药胶束,由两亲嵌段共聚物和疏水药物组成,所述疏水药物包埋在两亲嵌段共聚物形成的疏水内核中,所述两亲嵌段共聚物的结构式如下:
其中,R为C2~C12的烷基,x=45~113,y/(y+z)=0.09~0.13,y+z=50~210。
上述K+响应型两亲嵌段共聚物载药胶束,所述载药胶束中疏水药物的含量为3~10wt%。
上述K+响应型两亲嵌段共聚物载药胶束,所述载药胶束粒径为70~200nm。
本发明所述K+响应型两亲嵌段共聚物载药胶束的制备方法,工艺步骤如下:
(1)将两亲嵌段共聚物和疏水性药物溶于所述两亲嵌段共聚物和疏水性药物的共溶剂中配制成溶液,共溶剂的量以两亲嵌段共聚物和疏水药物能完全溶解为限;所述两亲嵌段共聚物的结构式如下
其中,R为C2~C12的烷基,x=45~113,y/(y+z)=0.09~0.13,y+z=50~210;
(2)将步骤(1)所得溶液加热至高于两亲嵌段共聚物的低临界溶解温度,在搅拌下向溶液中滴加去离子水或蒸馏水得到两亲嵌段共聚物浓度为0.1~2mg/ml的混合液,然后于与溶液温度相同的去离子水或蒸馏水中透析除去所述共溶剂,过滤透析后的混合液除去残留杂质以及细菌,即得K+响应型两亲嵌段共聚物载药胶束溶液,经冷冻干燥得到K+响应型两亲嵌段共聚物载药胶束;
或者将步骤(1)所得溶液在搅拌下滴加入温度高于两亲嵌段共聚物的低临界溶解温度的去离子水或蒸馏水中得到两亲嵌段共聚物浓度为0.1~2mg/ml的混合液,然后于相同温度的去离子水或蒸馏水中透析除去所述共溶剂,过滤透析后的混合液除去残留杂质以及细菌,即得 K+响应型两亲嵌段共聚物载药胶束溶液,经冷冻干燥得到K+响应型两亲嵌段共聚物载药胶束。
上述K+响应型两亲嵌段共聚物载药胶束的制备方法,所述两亲嵌段共聚物的制备方法如下:
(1)大分子链转移剂的合成
将聚乙二醇单甲醚、2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸、4-二甲氨基吡啶和二氯甲烷加入反应容器中并冷却至0℃,搅拌至混合均匀形成混合液,然后将二环己基碳二亚胺溶解在二氯甲烷中后加入所得混合液中形成反应体系,并于室温下搅拌反应24~72h,反应结束后将所得反应液过滤除去沉淀物,将滤液浓缩后滴加到无水乙醚中沉淀后过滤,用四氢呋喃溶解过滤所得滤饼后再用无水乙醚沉淀,过滤,将过滤所得滤饼干燥,即得大分子链转移剂;
所述聚乙二醇单甲醚、2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸、二环己基碳二亚胺的摩尔比为1:(1.25~2.35):(2.1~2.85),二氯甲烷的使用量应使反应体系中聚乙二醇单甲醚、 2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸和二环己基碳二亚胺的总浓度为10wt%~15wt%; 4-二甲氨基吡啶的用量为聚乙二醇单甲醚质量的0.6%~0.8%;
(2)两亲嵌段共聚物的合成
将步骤(1)所得大分子链转移剂、N-异丙基丙烯酰胺、苯并18冠6丙烯酰胺、偶氮二异丁腈和1,4-二氧六环加入反应容器中形成混合液,向反应容器的液面下通入氮气以除去所得混合液中的氧气,然后在氮气保护下于65℃~80℃反应12~36h,反应结束后将所得反应液用四氢呋喃稀释后滴加到无水乙醚中沉淀后过滤,将过滤所得滤饼用四氢呋喃溶解后再用无水乙醚沉淀,过滤,将过滤所得滤饼干燥,即得两亲嵌段共聚物聚(乙二醇)单甲醚-嵌段 -聚(N-异丙基丙烯酰胺-共聚-苯并18冠6丙烯酰胺);
步骤(1)所得大分子链转移剂、N-异丙基丙烯酰胺、偶氮二异丁腈的摩尔比为1: (100~200):(0.05~0.5),所述苯并18冠6丙烯酰胺的摩尔量占苯并18冠6丙烯酰胺和N- 异丙基丙烯酰胺摩尔量之和的9%~13%,1,4-二氧六环的使用量应使混合液中N-异丙基丙烯酰胺的浓度为10~30wt%。
上述K+响应型两亲嵌段共聚物载药胶束的制备方法,所述共溶剂只要能将两亲嵌段共聚物的疏水嵌段与亲水嵌段以及疏水性药物良好溶解即可,具体会因疏水性药物的不同而有所不同,所述共溶剂可选用四氢呋喃、N,N-二甲基甲酰胺、1,4-二氧六环中的一种。
上述K+响应型两亲嵌段共聚物载药胶束的制备方法,步骤(2)中,向溶液中滴加去离子水或蒸馏水的滴加速率为10~20s/滴,将溶液滴加入去离子水或蒸馏水的滴加速率为10~20 s/滴,透析除去共溶剂时每2~5h换一次水,透析时间为5~7天,过滤透析后的混合液时采用滤膜,滤膜的滤膜孔径为0.25~0.45μm。
上述K+响应型两亲嵌段共聚物载药胶束的制备方法,步骤(1)中,疏水性药物的投加量与载药胶束的载药量有关,疏水性药物的投加量根据实际应用中对载药量的需求进行确定,疏水药物的投料量为两亲嵌段共聚物质量的10~20%。
上述K+响应型两亲嵌段共聚物载药胶束的制备方法,疏水性药物根据实际应用需求进行确定,可以为泼尼松龙、姜黄素、吲哚美辛中的一种。
本发明所述K+响应型两亲嵌段共聚物载药胶束的药物释放机理如下:
两亲嵌段共聚物通过可逆加成-断裂链转移自由基聚合(RAFT)合成,以生物相容性良好的聚(乙二醇)单甲醚(PEG)作为胶束的亲水外壳嵌段,以N-异丙基丙烯酰胺(NIPAM) 和苯并18冠6丙烯酰胺(B18C6Am)的共聚物作为胶束的疏水内核嵌段。该两亲嵌段共聚物中的18冠6能够选择性络合K+,在络合K+后,两亲嵌段共聚物的亲水性增加,其LSCT 向高温迁移。本发明所述载药胶束是两亲嵌段共聚物与疏水性药物自组装形成的胶束,疏水性的药物包裹在胶束内核之中。载药胶束进入血液循环后,由于胞外的K+浓度(3.5-5.5 mmol/L)较低,而胶束对低浓度K+响应性较小,载药胶束的LCST低于体温,体温对其形态不会产生太大的影响,载药胶束仍保持良好的胶束形态。当胶束被细胞內吞进入细胞内,因细胞内部的K+浓度(140-150mmol/L)较高,两亲嵌段共聚物特异性地络合钾离子,使得胶束疏水内核的疏水能力下降,亲水能力增强,载药胶束的LCST高于体温,其疏水内核变得亲水,胶束的亲疏水平衡被打破,胶束解离,从而释放包载的药物。
与现有技术相比,本发明具有以下有益效果:
1、本发明提供了一种新的K+响应型两亲嵌段共聚物载药胶束,丰富了该类型载药胶束的种类。
2.本发明所述载药胶束在响应胞内K+浓度条件后,胶束解离,释放所包载的药物,此时,由于18冠6特异性络合K+,两亲嵌段共聚物的亲水性增加,LSCT向高温迁移,作为疏水内核的疏水嵌段转变为亲水嵌段,整个两亲嵌段共聚物转变为亲水性的高分子。与现有的K+响应型胶束相比,由于该两亲嵌段共聚物释药后转变为亲水的高分子,能溶解于体内,且其相对分子质量足够小(<40000),因此能被肾脏清除,可经正常机体代谢途径排出体外,从而避免两亲嵌段共聚物在体内蓄积引发潜在的毒性。
3.本发明所述K+响应型两亲嵌段共聚物载药胶束由于两亲嵌段共聚物中引入了18冠6,由于18冠6是一种能选择性络合K+的冠醚,具有特异性络合K+的特点,可实现载药胶束在细胞内外不同K+浓度条件下对两亲嵌段共聚物的LCST的调节,从而实现打破胶束的亲疏水平衡,胶束解离,释放所包载的药物的目的。嵌段共聚物的构建过程中不需要另外添加亲水或疏水的物质来特别调节整个嵌段的LCST,其结构中原本的18冠6就能完成所需LCST的转变,使嵌段共聚物结构构建容易,合成步骤简单。
4、本发明所述载药胶束中所使用的亲水外壳材料是PEG,其生物相容性好,而且能避免 RES的识别和清除,进一步延长胶束在血液中的循环时间,提高药物的利用率。
附图说明
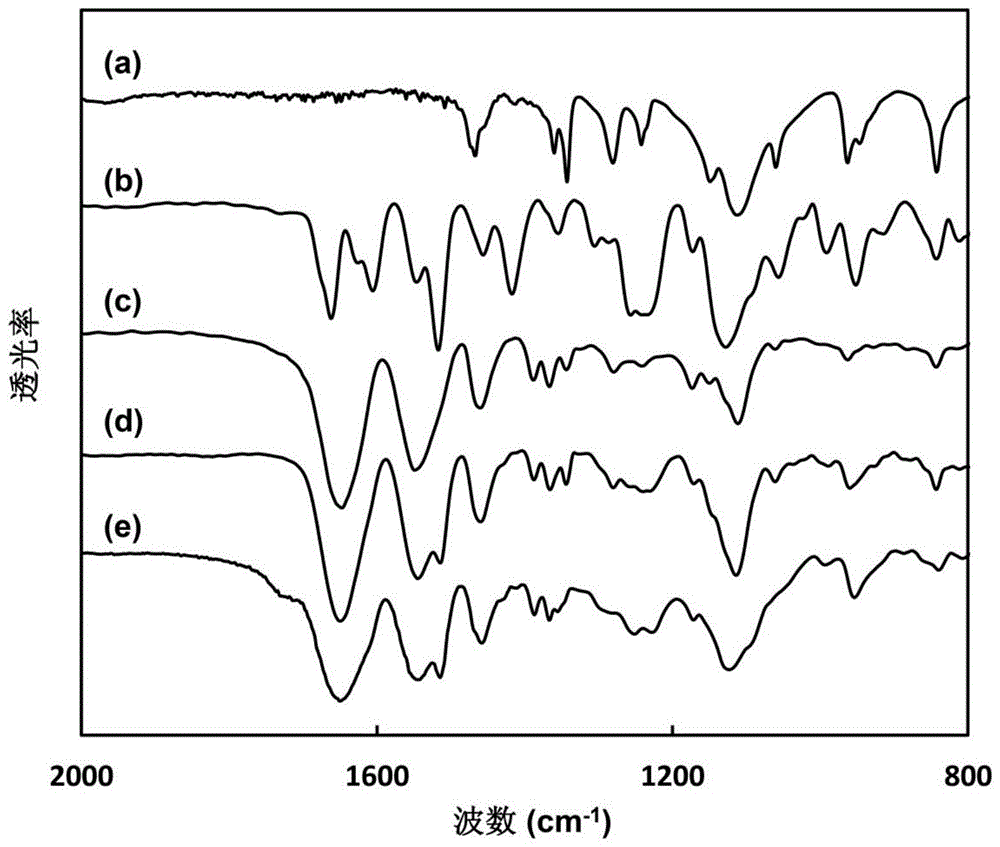
图1中的(a)(b)(c)(d)(e)分别为PEG-CTA、B18C6Am、对比例1制备的PEG-b-PNIPAM、对比例2制备的PEG-b-P(NIPAM-co-B18C6Am)和实施例1制备的PEG-b-P(NIPAM-co- B18C6Am)的红外图谱。
图2中的(a)(b)(c)(d)分别为PEG-CTA、对比例1制备的PEG-b-PNIPAM、对比例2制备的PEG-b-P(NIPAM-co-B18C6Am)和实施例1制备的PEG-b-P(NIPAM-co-B18C6Am) 的核磁图谱。
图3为对比例1制备的PEG-b-PNIPAM、对比例2制备的PEG-b-P(NIPAM-co-B18C6Am) 和实施例1制备的PEG-b-P(NIPAM-co-B18C6Am)在去离子水、细胞内液模拟液和细胞外液模拟液中的低临界溶解温度(LCST)。
图4为实施例1制备的PEG-b-P(NIPAM-co-B18C6Am)以芘为荧光探针得到的荧光激发光谱的I383.4/I372.8随浓度的变化。
图5(a)(b)分别为实施例1制备的空白胶束与载药胶束的粒径分布图。
图6(a)(b)分别为实施例1制备的空白胶束与载药胶束的透射电镜(TEM)图。
图7为实施例1中载药胶束在细胞内液模拟液和细胞外液模拟液中的药物释放图。
具体实施方式
以下通过实施例对本发明所述K+响应型两亲嵌段共聚物载药胶束及其制备方法作进一步说明。
下述各实施例中,所述苯并18冠6丙烯酰胺(B18C6Am)参照R.Ungaro,B.E.Haj,J.Smid, J.Am.Chem.Soc.,1976,98,5198和K.Yagi,J.A.Ruiz,M.C.Sanchez,Makromol.Chem.,Rapid Commun.,1980,1:263-268中的方法自行合成;所述2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸购自sigma;所述聚乙二醇单甲醚(PEG)购自sigma;所述N-异丙基丙烯酰胺(NIPAM)使用前用正己烷/丙酮重结晶,偶氮二异丁腈(AIBN)使用前用乙醇重结晶。
所述红外分光光度计型号为IRPrestige-21,核磁共振型号为VNMR 6.1C,紫外分光光度计型号为岛津UV-1700,荧光分光光度计型号为RF-5301PC,动态光散射分光光度计型号为 ZEN 3690,TEM型号为Tecnai G2F20S-TWIN。
实施例1
本实施例中,制备两亲嵌段共聚物—聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺- 共聚-苯并18冠6丙烯酰胺)(PEG-b-P(NIPAM-co-B18C6Am)),其结构式如下:
该结构式中,x=113,y=26,z=179;
(1)合成聚(乙二醇)单甲醚大分子链转移剂(PEG-CTA)
将聚乙二醇单甲醚(PEG)、三硫代碳酸酯、4-二甲氨基吡啶(DMAP)和溶剂二氯甲烷加入圆底烧瓶中并冷却至0℃,搅拌至混合均匀形成混合液,将二环己基碳二亚胺(DCC) 溶解于少量二氯甲烷中后缓慢滴加入上述混合液,室温下搅拌反应48h,将所得反应液过滤除去沉淀物,滤液浓缩后缓慢滴加到大量无水乙醚中沉淀,静置,过滤,得到白色滤饼,用适量四氢呋喃溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置,过滤,再如此溶解沉淀一次后,将所得产品于40℃下真空干燥,即得淡黄色的大分子链转移剂PEG-CTA;
所述聚乙二醇单甲醚、三硫代碳酸酯和二环己基碳二亚胺的摩尔比为1:2.35:2.85,二氯甲烷的使用量为混合液中聚乙二醇单甲醚、三硫代碳酸酯和二环己基碳二亚胺的总浓度为 11.5wt%;4-二甲氨基吡啶的用量为聚乙二醇单甲醚质量的0.75%。
(2)合成两亲嵌段共聚物
将步骤(1)所得大分子链转移剂PEG-CTA、N-异丙基丙烯酰胺(NIPAM)、苯并18冠 6丙烯酰胺(B18C6Am)、引发剂偶氮二异丁腈(AIBN)和溶剂1,4-二氧六环加入圆底烧瓶中形成混合液,在搅拌状态下向圆底烧瓶的液面下通入30min氮气以除去所述混合液中的氧气,在氮气保护下于70℃条件下反应24h,将所得反应液用四氢呋喃稀释后滴加到大量无水乙醚中沉淀后过滤得白色滤饼,然后用适量四氢呋喃溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置,过滤,再如此溶解沉淀一次后,将所得滤饼于40℃下真空干燥,即得白色固体两亲嵌段共聚物聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺-共聚-苯并18 冠6丙烯酰胺)(PEG-b-P(NIPAM-co-B18C6Am);
所述步骤(1)所得大分子链转移剂PEG-CTA、NIPAM、AIBN的摩尔比为1:200:0.2,所述B18C6Am的摩尔量占B18C6Am和NIPAM摩尔量之和的15%,1,4-二氧六环的使用量为使混合液中NIPAM的浓度为10wt%。
PEG-CTA、B18C6Am和本实施例制备的PEG-b-P(NIPAM-co-B18C6Am)的红外光谱分别如图1(a)、(b)、(e)所示,1370cm-1和1390cm-1的两个峰是NIPAM中的异丙基-CH(CH3)2中甲基的红外特征吸收峰,1500cm-1附近的吸收峰是B18C6Am中苯环骨架伸缩振动吸收峰, 1650cm-1处的强峰是NIPAM和B18C6Am中酰胺羰基的伸缩振动吸收峰,1100cm-1附近的吸收峰是PEG和B18C6Am中C-O醚键的红外吸收峰,以上所述特征峰均出现在 PEG-b-P(NIPAM-co-B18C6Am)的红外图谱中,说明本实施例成功制备得到两亲嵌段共聚物 PEG-b-P(NIPAM-co-B18C6Am)。
PEG-CTA和本实施例制备得到的PEG-b-P(NIPAM-co-B18C6Am)的核磁图谱如图2(a) (d)所示,从图中可以看到明显的PEG特征峰(3.76ppm,-OCH2CH2O-);NIPAM的特征峰(1.19ppm,-CH(CH3)2));B18C6Am的特征峰(6.5-7.5ppm,Ar-H;4.26ppm, ArOCH2CH2O-;3.94ppm,-OCH2CH2O-),以上核磁数据进一步说明本实施例成功合成了两亲嵌段共聚物PEG-b-P(NIPAM-co-B18C6Am)。
对比例1不加苯并18冠6丙烯酰胺
本对比例中,制备两亲嵌段共聚物—聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺 (PEG-b-PNIPAM),
该结构式中,x=113,z=179。
(1)合成大分子链转移剂(PEG-CTA)
将PEG、三硫代碳酸酯、DMAP和溶剂二氯甲烷加入圆底烧瓶中并冷却至0℃,搅拌至混合均匀形成混合液,将DCC溶解于少量二氯甲烷中后缓慢滴加入上述混合液,室温下搅拌反应48h,将所得反应液过滤除去沉淀物,滤液浓缩后缓慢滴加到大量无水乙醚中沉淀,静置过滤后得到白色滤饼,用适量四氢呋喃溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置过滤,再如此溶解沉淀一次后,将所得滤饼于40℃下真空干燥,即得淡黄色的大分子链转移剂PEG-CTA;
所述PEG、三硫代碳酸酯和DCC的摩尔比为1:2.35:2.85,二氯甲烷的使用量为混合液中PEG、三硫代碳酸酯和DCC的总浓度为11.5wt%;4-二甲氨基吡啶的用量为聚乙二醇单甲醚质量的0.75%。
(2)合成两亲嵌段共聚物PEG-b-PNIPAM
将步骤(1)所得大分子链转移剂PEG-CTA、NIPAM、引发剂AIBN和溶剂1,4-二氧六环加入圆底烧瓶中形成混合液,在搅拌状态下向圆底烧瓶的液面下通入30min氮气以除去所述混合液中的氧气,于70℃条件下反应24h,将所得反应液用四氢呋喃稀释后滴加到大量无水乙醚中沉淀后过滤得白色滤饼,然后用适量四氢呋喃溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置过滤,再如此溶解沉淀一次后,将所得滤饼于40℃下真空干燥,即得白色固体两亲嵌段共聚物聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺) (PEG-b-PNIPAM)。
所述步骤(1)所得大分子链转移剂PEG-CTA、NIPAM、AIBN的摩尔比为1:200:0.2,溶剂1,4-二氧六环的使用量为使混合液中NIPAM的浓度为10wt%。
PEG-CTA和本对比例制备的PEG-b-PNIPAM的红外光谱如图1 (a)、(c)所示。从图中可以看出,1370cm-1和1390cm-1两个强度为1:1的峰是NIPAM的异丙基-CH(CH3)2中甲基的对称面内弯曲振动在1380cm-1处分裂的红外特征吸收峰,1650cm-1处的峰是NIPAM中的酰胺羰基的伸缩振动峰,1100cm-1附近的吸收峰是PEG中C-O醚键的红外特征吸收峰,以上所述数据说明本对比例成功制备合成了PEG-b-PNIPAM。
PEG-CTA和本对比例制备的PEG-b-PNIPAM的核磁图谱如图2(a)、图2(b)所示,从图中可以看到明显的PEG特征峰(3.76ppm,-OCH2CH2O-),NIPAM的特征峰(1.19ppm, -CH(CH3)2)),以上所述核磁数据进一步说明本对比例成功合成了两亲嵌段共聚物 PEG-b-PNIPAM。
对比例2
本对比例中,制备两亲嵌段共聚物—聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺- 共聚-苯并18冠6丙烯酰胺)(PEG-b-P(NIPAM-co-B18C6Am),其结构式如下:
该结构式中,x=113,y=19,z=179。
(1)合成聚(乙二醇)单甲醚大分子链转移剂(PEG-CTA)
将PEG、三硫代碳酸酯、DMAP和溶剂二氯甲烷加入圆底烧瓶中并冷却至0℃,搅拌至混合均匀形成混合液,将DCC溶解于少量二氯甲烷中后缓慢滴加入上述混合液,室温下搅拌反应48h,将所得反应液过滤除去沉淀物,滤液减压浓缩后缓慢滴加到大量无水乙醚中沉淀,静置过滤后得到白色滤饼,用适量四氢呋喃溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置过滤,再如此溶解沉淀一次后,将所得滤饼于40℃下真空干燥,即得淡黄色的大分子链转移剂PEG-CTA;
所述PEG、三硫代碳酸酯和DCC的摩尔比为1:2.35:2.85,二氯甲烷的使用量为混合液中PEG、三硫代碳酸酯和DCC的总浓度为11.5wt%;4-二甲氨基吡啶的用量为聚乙二醇单甲醚质量的0.75%。
(2)合成两亲嵌段共聚物
将步骤(1)所得大分子链转移剂PEG-CTA、NIPAM、B18C6Am、引发剂AIBN和溶剂 1,4-二氧六环加入圆底烧瓶中形成混合液,在搅拌状态下向圆底烧瓶的液面下通入30min氮气以除去所述混合液中的氧气,除氧后于70℃条件下反应24h,将所得反应液用四氢呋喃稀释后滴加到大量无水乙醚中沉淀后过滤得白色滤饼,然后用适量四氢呋喃溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置过滤,再如此溶解沉淀一次后,将所得滤饼于40℃下真空干燥,即得白色固体两亲嵌段共聚物聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺-共聚-苯并18冠6丙烯酰胺)(PEG-b-P(NIPAM-co-B18C6Am));
所述步骤(1)所得大分子链转移剂PEG-CTA、NIPAM、AIBN的摩尔比为1:200:0.2,所述B18C6Am的摩尔量占B18C6Am和NIPAM摩尔量之和的10%,1,4-二氧六环的使用量为使混合液中NIPAM的浓度为10wt%。
PEG-CTA、B18C6Am和本对比例制备的PEG-b-P(NIPAM-co-B18C6Am)的红外光谱分别如图1(a)、(b)、(d)所示,1370cm-1和1390cm-1的两个峰是NIPAM中的异丙基-CH(CH3)2中甲基的红外特征吸收峰,1500cm-1附近的吸收峰是B18C6Am中苯环骨架伸缩振动吸收峰, 1650cm-1处的强峰是NIPAM和B18C6Am中酰胺羰基的伸缩振动吸收峰,1100cm-1附近的吸收峰是PEG和B18C6Am中C-O醚键的红外吸收峰,以上所述特征峰均出现在 PEG-b-P(NIPAM-co-B18C6Am)的红外图谱中,说明本对比例成功制备得到两亲嵌段共聚物 PEG-b-P(NIPAM-co-B18C6Am)。
PEG-CTA和本对比例制备得到的PEG-b-P(NIPAM-co-B18C6Am)的核磁图谱如图2(a) (c)所示,从图中可以看到明显的PEG特征峰(3.76ppm,-OCH2CH2O-);NIPAM的特征峰(1.19ppm,-CH(CH3)2));B18C6Am的特征峰(6.5-7.5ppm,Ar-H;4.26ppm, ArOCH2CH2O-;3.94ppm,-OCH2CH2O-),以上所述核磁数据进一步说明本对比例成功合成了两亲嵌段共聚物PEG-b-P(NIPAM-co-B18C6Am)。
实施例2:实施例1和对比例1~2制备的两亲嵌段共聚物的K+响应性测定
将K+浓度为150mmol/L、Na+浓度为5mmol/L的混合水溶液作为细胞内液模拟液,K+浓度为5mmol/L、Na+浓度为150mmol/L的混合水溶液作为细胞外液模拟液;测定K+响应性时两亲嵌段共聚物的浓度均为0.1wt%。
测定对比例1制备得到的两亲嵌段共聚物PEG-b-PNIPAM在去离子水、细胞内液模拟液以及细胞外液模拟液中的低临界溶解温度(LCST),结果如图3所示。由图可知,制备所得 PEG-b-PNIPAM具有较好的温敏性;其在水中的LCST与PNIPAM在水中的LCST(32℃) 相近,在细胞内液模拟液和细胞外液模拟液中的LCST比在水中的LCST稍低,但两者结果相等,说明对比例1制备所得PEG-b-PNIPAM无K+响应性。
测定对比例2制备得到的两亲嵌段共聚物PEG-b-P(NIPAM-co-B18C6Am)在去离子水、细胞内液模拟液以及细胞外液模拟液中的LCST,结果如图3所示。由图可知,对比例2制备所得PEG-b-P(NIPAM-co-B18C6Am)具有较好的温敏性;其在细胞内液模拟液中的LCST比在细胞外液模拟液中的LCST高3.5℃,说明对比例2制备所得PEG-b-P(NIPAM-co-B18C6Am) 具有明显的K+响应性,与对比例1的K+响应性结果相比,也说明了两亲嵌段共聚物的K+响应性是由其中的B18C6Am所决定。
测定实施例1制备得到的两亲嵌段共聚物PEG-b-P(NIPAM-co-B18C6Am)在去离子水、细胞内液模拟液以及细胞外液模拟液中的LCST,结果如图3所示。由图可知,实施例1制备所得PEG-b-P(NIPAM-co-B18C6Am)具有较好的温敏性;其在细胞内液模拟液中的LCST比在细胞外液模拟液中的LCST高6.1℃,且与对比例2相比,其冠醚含量大于对比例2的冠醚含量,因此LCST的迁移程度更大,具有更加明显的K+响应性,更适于构建在生理条件下具有钾离子响应性的两亲嵌段共聚物胶束。
上述测定结果表明,以实施例1、对比例2制备所得PEG-b-P(NIPAM-co-B18C6Am)制备载药胶束,由于其在细胞外液模拟液中的LCST低于37℃,所以在细胞外液保持胶束形态,经过细胞內吞进入细胞后,由于其在细胞内液模拟液中的LCST高于37℃,使得胶束的疏水内核变得亲水,从而打破胶束的亲疏水平衡,胶束解离,释放所包载的药物。
实施例3:实施例1制备的两亲嵌段共聚物临界胶束浓度测定
测定实施例1制备所得两亲嵌段共聚物PEG-b-P(NIPAM-co-B18C6Am)的临界胶束浓度采用荧光法,以芘作为荧光探针测定,结果如图4所示。图中以两亲嵌段共聚物浓度的对数 (lgC)为X轴,I3/I1(I3为383.4nm处的荧光强度I383.4,I1为372.8nm处的荧光强度I372.8) 为Y轴,数据点的水平切线与变形曲线切线交点所对应的浓度即两亲嵌段共聚物的临界胶束浓度,为40mg/L,两亲嵌段共聚物浓度在此临界胶束浓度之上即可形成胶束。
实施例4:以实施例1制备的两亲嵌段共聚物制备空白胶束和载药胶束
1、制备空白胶束
(1)将实施例1制备所得两亲嵌段共聚物PEG-b-P(NIPAM-co-B18C6Am)充分溶解于少量N,N-二甲基甲酰胺(DMF)(使两亲嵌段共聚物和疏水药物溶解即可);
(2)将步骤(1)所得溶液加热至45℃,在剧烈搅拌下向所得溶液中缓慢滴加去离子水,去离子水滴加完毕后继续搅拌2h,其中去离子水的滴加速率为10s/滴,去离子水的滴加量为使PEG-b-P(NIPAM-co-B18C6Am)在最终所得溶液中的浓度为1mg/ml。将所得溶液转移至分子截留量为3500g/mol的透析袋中,在45℃条件下于去离子水中透析,每4h换一次水,透析5天,以除去有机溶剂。透析完成后,用滤膜孔径为0.45μm的过滤器过滤除菌,得到空白胶束溶液。该空白胶束的粒径分布如图5(a)所示,平均粒径为145.2nm,分布系数(PDI) 为0.131,其透射电镜(TEM)图片如图6(a)所示,由图可知,空白胶束具有均一的形态和较好的球形度。
2、制备载药胶束
(1)将疏水药物醋酸泼尼松龙以及实施例1制备得到的两亲嵌段共聚物 PEG-b-P(NIPAM-co-B18C6Am)(醋酸泼尼松龙:两亲嵌段共聚物=10%,w/w)充分溶解于少量DMF;
(2)将步骤(1)所得加热至45℃,在剧烈搅拌下向步骤(1)所得混合溶液中缓慢滴加去离子水,去离子水滴加完毕后继续搅拌溶液2h,其中,去离子水的滴加速率为10s/滴,去离子水的滴加量为使得PEG-b-P(NIPAM-co-B18C6Am)在最终所得溶液中的的浓度为1 mg/ml。将所得溶液转移至分子截留量为3500g/mol的透析袋中,在45℃条件下于去离子水中透析,每4h换一次水,透析5天,以除去有机溶剂和游离药物。透析完成后,用滤膜孔径为0.45μm的过滤器过滤除菌,得到载药胶束溶液。该载药胶束的粒径分布如图5(b)所示,平均粒径为163.9nm,分布系数(PDI)为0.163。载药胶束的透射电镜(TEM)图片如图6所示,由图可知,空白胶束呈较为均一的球形形态。
实施例5:实施例4制备的载药胶束的体外药物释放试验
本实施例中,将K+浓度为150mmol/L、Na+浓度为5mmol/L的混合水溶液作为细胞内液模拟液,K+浓度为5mmol/L、Na+浓度为150mmol/L的混合水溶液作为细胞外液模拟液,进行载药胶束体外模拟药物释放试验。
量取2ml实施例4所制备得到的载药胶束溶液,另用2ml的K+浓度为300mmol/L、Na+浓度为10mmol/L的盐溶液将2ml的胶束溶液稀释为K+浓度为150mmol/L、Na+浓度为5 mmol/L的载药胶束溶液,然后将得到的载药胶束溶液转移至分子截留量为3500g/mol的透析袋,37℃恒温下于细胞内液模拟液的水溶液中进行透析,在特定时间取透析外液测定药物在最大紫外吸收波长处的吸光度,计算醋酸泼尼松龙的累积释放量。
量取2ml实施例4所制备得到的载药胶束溶液,另用2ml的K+浓度为10mmol/L、Na+浓度为300mmol/L的盐溶液将2ml的胶束溶液稀释为K+浓度为5mmol/L、Na+浓度为150 mmol/L的载药胶束溶液,然后将得到的载药胶束溶液转移至分子截留量为3500g/mol的透析袋,于37℃细胞内液模拟液的水溶液中进行透析,在特定时间取透析外液测定药物在最大紫外吸收波长处的吸光度,计算醋酸泼尼松龙的累积释放量。
药物释放完成后,将透析袋内的剩余溶液冻干后用DMF溶解,测定药物在最大紫外吸收波长处的吸光度,计算载药胶束内部剩余的药物量。药物负载量按照以下公式计算,得出药物负载量为4.7%。
载药胶束在细胞内液模拟液与细胞外液模拟液中的药物释放行为如图7所示,由图可知,药物在细胞内液模拟液中的累计释放量明显高于在细胞外液模拟液中的累计释放量,说明K+的存在极大地影响了载药胶束的药物释放,制备得到的载药胶束表现出了良好的K+识别响应特性,能够实现对包载药物的控制释放。
实施例6
本实施例提供载药胶束的制备方法,步骤如下:
(1)制备两亲嵌段共聚物—聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺-共聚-苯并18冠6丙烯酰胺)(PEG-b-P(NIPAM-co-B18C6Am),其结构式如下:
该结构式中,x=113,y=22,z=180。
1)合成聚(乙二醇)单甲醚大分子链转移剂(PEG-CTA)
将聚乙二醇单甲醚(PEG)、三硫代碳酸酯、4-二甲氨基吡啶(DMAP)和溶剂二氯甲烷加入圆底烧瓶中并冷却至0℃,搅拌至混合均匀形成混合液,将二环己基碳二亚胺(DCC) 溶解于少量二氯甲烷中后缓慢滴加入上述混合液,室温下搅拌反应48h,将所得反应液过滤除去沉淀物,滤液浓缩后缓慢滴加到大量无水乙醚中沉淀,静置,过滤,得到白色滤饼,用适量THF溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置,过滤,再如此溶解沉淀一次后,将所得产品于40℃下真空干燥,即得淡黄色的大分子链转移剂PEG-CTA;
所述聚乙二醇单甲醚、三硫代碳酸酯和二环己基碳二亚胺的摩尔比为1:1.25:2.1,二氯甲烷的使用量为混合液中聚乙二醇单甲醚、三硫代碳酸酯和二环己基碳二亚胺的总浓度为 10wt%;4-二甲氨基吡啶的用量为聚乙二醇单甲醚质量的0.75wt%。
2)合成两亲嵌段共聚物
将步骤1)所得大分子链转移剂PEG-CTA、N-异丙基丙烯酰胺(NIPAM)、苯并18冠6 丙烯酰胺(B18C6Am)、引发剂偶氮二异丁腈(AIBN)和溶剂1,4-二氧六环加入圆底烧瓶中形成混合液,在搅拌状态下向圆底烧瓶的液面下通入30min氮气以除去所述混合液中的氧气,于70℃条件下反应24h,将所得反应液用四氢呋喃稀释后滴加到大量无水乙醚中沉淀后过滤得白色滤饼,然后用适量THF溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置,过滤,再如此溶解沉淀一次后,将所得产品于40℃下真空干燥,即得白色固体两亲嵌段共聚物聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺-共聚-苯并18冠6丙烯酰胺) (PEG-b-P(NIPAM-co-B18C6Am));
所述步骤所得大分子链转移剂PEG-CTA、NIPAM、AIBN的摩尔比为1:100:0.2,所述B18C6Am的摩尔量占B18C6Am和NIPAM摩尔量之和的10.9%,1,4-二氧六环的使用量为使混合液中NIPAM的浓度为15wt%。
(2)将疏水药物吲哚美辛以及步骤(1)所制备的两亲嵌段共聚物PEG-b-P(NIPAM-co- B18C6Am)(吲哚美辛:两亲嵌段共聚物=15%,w/w)溶解于1,4-二氧六环中;
(3)将步骤(2)所得溶液置于45℃温度条件下,在剧烈搅拌下向溶液中以10s/滴的速率滴加去离子水,去离子水滴加完毕后继续搅拌溶液2h,去离子水的滴加量为使得 PEG-b-P(NIPAM-co-B18C6Am)在最终所得溶液中的浓度为0.5mg/ml。将所得溶液转移至分子截留量为3500g/mol的透析袋中,在45℃条件下于去离子水中透析,每4h换一次水,透析5天,以除去有机溶剂和游离药物。透析完成后,用滤膜孔径为0.45μm的过滤器过滤除菌,得到载药胶束溶液。
实施例7
本实施例提供载药胶束的制备方法,步骤如下:
(1)制备两亲嵌段共聚物—聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺-共聚-苯并18冠6丙烯酰胺)(PEG-b-P(NIPAM-co-B18C6Am),其结构式如下:
该结构式中,x=50,y=10,z=78。
1)合成聚(乙二醇)单甲醚大分子链转移剂(PEG-CTA)
将聚乙二醇单甲醚(PEG)、三硫代碳酸酯、4-二甲氨基吡啶(DMAP)和溶剂二氯甲烷加入圆底烧瓶中并冷却至0℃,搅拌至混合均匀形成混合液,将二环己基碳二亚胺(DCC) 溶解于少量二氯甲烷中后缓慢滴加入上述混合液,室温下搅拌反应72h,将所得反应液过滤除去沉淀物,滤液浓缩后缓慢滴加到大量无水乙醚中沉淀,静置,过滤,得到白色滤饼,用适量THF溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置,过滤,再如此溶解沉淀一次后,将所得产品于40℃下真空干燥,即得淡黄色的大分子链转移剂PEG-CTA;
所述聚乙二醇单甲醚、三硫代碳酸酯和二环己基碳二亚胺的摩尔比为1:1.5:2.35,二氯甲烷的使用量为混合液中聚乙二醇单甲醚、三硫代碳酸酯和二环己基碳二亚胺的总浓度为 15wt%;4-二甲氨基吡啶的用量为聚乙二醇单甲醚质量的0.75%。
2)合成两亲嵌段共聚物
将步骤1)所得大分子链转移剂PEG-CTA、N-异丙基丙烯酰胺(NIPAM)、苯并18冠6 丙烯酰胺(B18C6Am)、引发剂偶氮二异丁腈(AIBN)和溶剂1,4-二氧六环加入圆底烧瓶中形成混合液,在搅拌状态下向圆底烧瓶的液面下通入20min氮气以除去所述混合液中的氧气,于75℃条件下反应24h,将所得反应液用四氢呋喃稀释后滴加到大量无水乙醚中沉淀后过滤得白色滤饼,然后用适量THF溶解滤饼,在搅拌状态下将其缓慢滴入大量乙醚中,静置,过滤,再如此溶解沉淀一次后,将所得产品于40℃下真空干燥,即得白色固体两亲嵌段共聚物聚(乙二醇)单甲醚-嵌段-聚(N-异丙基丙烯酰胺-共聚-苯并18冠6丙烯酰胺) (PEG-b-P(NIPAM-co-B18C6Am));
所述步骤1)所得大分子链转移剂PEG-CTA、NIPAM、AIBN的摩尔比为1:100:0.4,所述B18C6Am的摩尔量占B18C6Am和NIPAM摩尔量之和的12.5%,1,4-二氧六环的使用量为使混合液中NIPAM的浓度为25wt%。
(2)将疏水药物姜黄素以及步骤(1)所制备的两亲嵌段共聚物PEG-b-P(NIPAM-co- B18C6Am)(姜黄素:两亲嵌段共聚物=18%,w/w)溶解于THF中;
(3)将步骤(2)所得溶液置于45℃温度条件下,在剧烈搅拌下向混合溶液中以20s/ 滴的速率滴加去离子水,去离子水滴加完毕后继续搅拌溶液2h,去离子水的滴加量为使得 PEG-b-P(NIPAM-co-B18C6Am)在最终所得溶液中的浓度为2mg/ml。将所得溶液转移至分子截留量为3500g/mol的透析袋中,在45℃条件下于去离子水中透析,每4h换一次水,透析 5天,以除去有机溶剂和游离药物。透析完成后,用滤膜孔径为0.45μm的过滤器过滤除菌,得到载药胶束溶液。
- 还没有人留言评论。精彩留言会获得点赞!