一种新的阿莫西林颗粒的制作方法
本发明涉及一种颗粒剂及其制备方法,尤其涉及一种新的阿莫西林颗粒及其制备方法,属于医药
技术领域:
。
背景技术:
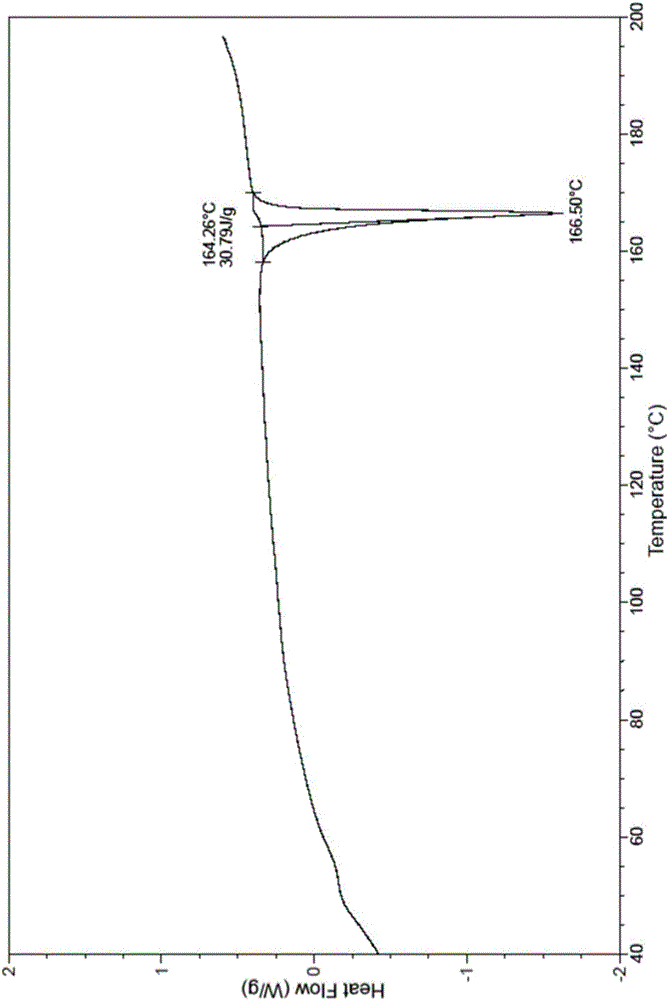
:阿莫西林(Amoxicillin),又名羟氨苄青霉素,其化学名称为(2S,5R,6R)-3,3-二甲基-6-[(R)-(-)-2-氨基-2-(4-羟基苯基)乙酰氨基]-7-氧代-4-硫杂-1-氮杂双环[3.2.0]庚烷-2-甲酸三水合物,分子式为C16H19N3O5S·3H2O,分子量为419.46。阿莫西林的性状为类白色或白色粉末,呈结晶状,味微苦,不溶于乙醇,微溶于水,其化学结构式为:阿莫西林是半合成青霉素类抗生素,对革兰阳性菌和某些革兰阴性菌都具有良好的抗菌作用。阿莫西林在体内抗菌作用明显优于氨苄青霉索,其抗菌活性起主要作用的基本结构是6-氨基青霉烷酸中的β-内酰胺环,可专一性地与细菌内膜上靶位点结合,抑制细菌细胞壁黏肽合成酶的活性,从而阻碍细胞壁黏肽的合成,使细菌的细胞壁缺损,菌体膨胀裂解。阿莫西林是目前应用较为广泛的口服青霉素之一,其制剂有胶囊、片剂、颗粒剂、分散片等等。目前市场上的阿莫西林颗粒大多采用传统生产工艺,采用湿法制粒,而阿莫西林原料对湿热非常敏感,传统的湿法制粒生产工艺对产品质量产生影响,含量下降明显,有关物质尤其是高分子杂质增加显著,产品稳定性差。而已有研究表明,阿莫西林自身聚合形成不同聚合度的混合物,是引发过敏反应的主要原因。控制阿莫西林制剂中的高分子杂质含量,是减少青霉素类抗生素过敏反应、提高药物安全性的根本途径。因而目前市售阿莫西林颗粒存在杂质较高、稳定性差等问题。专利CN201010556659.7涉及一种阿莫西林颗粒及其制备工艺,该颗粒制备工艺中采用低水分、易挥发的黏合剂溶液和低温干燥技术,避免温湿度对产品质量的影响,产品质量和稳定性得到一定改善,但对杂质的控制还是不够理想,会增加药物不良反应的发生。因此,临床上急需开发出一种更稳定、更安全的阿莫西林颗粒产品。技术实现要素:本发明目的在于,针对现有技术的缺陷,提供了一种新的阿莫西林颗粒,该阿莫西林颗粒的活性成分是一种不同于现有技术的新晶型的阿莫西林化合物,并通过实验证实,该种晶型的阿莫西林化合物的稳定性优于已有晶型,杂质含量低,且能保持药物长时间放置后含量下降很少,有利于制剂的长期稳定,从而保证了用药的安全性。为实现本发明目的,采用以下技术方案:本发明提供一种新的阿莫西林颗粒,该新的阿莫西林颗粒由以下重量份的组分制备而成:阿莫西林125g~250g、碳酸氢钠6300g~10800g、乙交酯丙交酯共聚物(34.7:65.3)0.10g~0.50g、羧甲基淀粉钠120g~260g、硬脂富马酸钠130g~280g、甘露醇1500g~3250g、乳糖1520g~3090g、蔗糖13000g~26500g、淀粉12500g~25800g、聚维酮K3030g~56g、甜橙香精108g~220g、安赛蜜116g~220g;其中,所述阿莫西林为新晶型的阿莫西林,如式(I)所示:使用Cu-Ka射线测量得到的X-射线粉末衍射图谱中在2θ为11.62±0.1°、16.29±0.1°、17.51±0.1°、20.04±0.1°、20.80±0.1°、22.32±0.1°、23.01±0.1°、27.56±0.1°处显示特征峰。上述一种新的阿莫西林颗粒,所述新晶型的阿莫西林的差示扫描量热图谱在163℃-168℃有一个吸热峰。上述一种新的阿莫西林颗粒,所述新晶型的阿莫西林,使用Cu-Ka射线测量得到的X-射线粉末衍射图谱中在2θ为5.78±0.1°、11.82±0.1°、12.57±0.1°、13.78±0.1°、15.73±0.1°、16.84±0.1°、18.81±0.1°、19.28±0.1°、23.44±0.1°、24.69±0.1°、25.08±0.1°、27.80±0.1°、28.82±0.1°、33.06±0.1°、33.71±0.1°处显示特征峰。上述一种新的阿莫西林颗粒,所述新晶型的阿莫西林的差示扫描量热图谱在166.50±2℃处有吸热峰。根据前述的新的阿莫西林颗粒,其中,所述新晶型的阿莫西林的制备方法包括以下步骤:将阿莫西林粗品加入一定配比的甲醇与N-甲基吡咯烷酮的混合溶剂中,加热至回流溶解,再向溶液中加入无水乙醇进而形成混合溶剂体系,然后降温冷却析晶,过滤,洗涤,真空干燥,得到新晶型的阿莫西林。上述新的阿莫西林颗粒,所述阿莫西林粗品与甲醇和N-甲基吡咯烷酮的重量g:体积ml:体积ml之比为10:65~75:12~16。上述新的阿莫西林颗粒,所述混合溶剂体系中甲醇、N-甲基吡咯烷酮和无水乙醇的体积ml比为65~75:12~16:6~10。上述新的阿莫西林颗粒,所述的降温冷却析晶,其温度降为0~15℃。上述新的阿莫西林颗粒,所述的洗涤,其使用的溶剂为石油醚。上述新的阿莫西林颗粒,所述的真空干燥,其干燥所使用的温度40~55℃。本发明还提供了一种上述新的阿莫西林颗粒的制备方法,包括如下步骤:将处方量上述新晶型阿莫西林化合物原料药过100目筛,辅料过40目筛,备用;按处方重量配比称取处方量的阿莫西林、56%处方量的碳酸氢钠、47%处方量的乙交酯丙交酯共聚物(34.7:65.3)和40%处方量的硬脂富马酸钠,混合均匀,干法制粒,制成细颗粒;加入处方量的羧甲基淀粉钠和剩余处方量的碳酸氢钠及乙交酯丙交酯共聚物(34.7:65.3),混合均匀,干法制粒,制成颗粒;将上述颗粒再与剩余处方量的硬脂富马酸钠、处方内其他辅料混匀后,装袋,即得。有文献报道,通过改变晶型可提高制剂稳定性,目前针对阿莫西林的晶型已有了一定的研究,如专利CN201610060313.5提供了一种新晶型阿莫西林化合物制备得到的胶囊剂稳定性更好,用药安全有效。在晶体领域,某种化合物是否存在晶体形式、存在多少种晶体形式以及存在何种晶体形式是不可预期的,因此,化合物晶体的发明或其制备带有不可预期性,某种新晶型的获得一般需要依赖实验结果加以确定。本发明人通过大量实验,通过对结晶工艺参数的严格控制,意外的获得了与现有技术均不同的新晶型阿莫西林。通过本发明实施例1-3可以看出,本发明所制备得到的新阿莫西林流动性好,更能满足药物制剂学的要求,更适合制备各种药物制剂。通过本发明实施例5-7可以看出,由本发明所述的新晶型阿莫西林加之添加入新型稳定剂辅料乙交酯丙交酯共聚物(34.7:65.3)及其辅料配比制备得到的颗粒剂稳定性好,从而提高了用药安全性和有效性,避免不良反应的发生率。附图说明图1本发明实施例1所述新晶型的阿莫西林的X-射线粉末衍射图谱图2本发明实施例1所述新晶型的阿莫西林的DSC谱图具体实施方式下面通过具体实施方式对本发明作进一步详细说明,但只是用于帮助理解本发明,使本领域专业技术人员能够实现或使用本发明,不对本发明构成任何限制。实施例1制备本发明所述的新晶型的阿莫西林将阿莫西林粗品1000g加入25L三口瓶内,加甲醇7000mL和N-甲基吡咯烷酮1400mL,搅拌、加热至回流溶解,再向溶液中加入800mL无水乙醇,然后缓慢降温冷却至5℃,搅拌析晶,过滤,石油醚洗涤,50℃真空干燥,得到新晶型的阿莫西林977g,收率97.7%。该新晶型的阿莫西林的X-射线粉末衍射图在反射角2θ为(精度为±0.1°):5.78°、11.62°、11.82°、12.57°、13.78°、15.73°、16.29°、16.84°、17.51°、18.81°、19.28°、20.04°、20.80°、22.32°、23.01°、23.44°、24.69°、25.08°、27.56°、27.80°、28.82°、33.06°、33.71°处有特征吸收峰,如图1所示。其DSC图谱在166.50℃附近有吸热峰(精度为±2℃),如图2所示。实施例2制备本发明所述的新晶型的阿莫西林将阿莫西林粗品1000g加入25L三口瓶内,加甲醇6500mL和N-甲基吡咯烷酮1200mL,搅拌、加热至回流溶解,再向溶液中加入600mL无水乙醇,然后缓慢降温冷却至15℃,搅拌析晶,过滤,石油醚洗涤,40℃真空干燥,得到新晶型的阿莫西林936g,收率93.6%。实施例3制备本发明所述的新晶型的阿莫西林将阿莫西林粗品1000g加入25L三口瓶内,加甲醇7500mL和N-甲基吡咯烷酮1600mL,搅拌、加热至回流溶解,再向溶液中加入1000mL无水乙醇,然后缓慢降温冷却至0℃,搅拌析晶,过滤,石油醚洗涤,55℃真空干燥,得到新晶型的阿莫西林959g,收率95.9%。实施例4制备本发明所述的乙交酯丙交酯共聚物(PLGA)将乙交酯(GA)与L-丙交酯(LLA)按照一定重量比例放入三口烧瓶中,加入催化剂辛酸亚锡(乙交酯单体质量的0.78%)和引发剂十二烷醇(乙交酯单体质量的0.33%),抽真空至10Pa,在搅拌条件下加热至185℃,反应6h,停止搅拌,维持真空度保温8h,然后自然冷却到室温。用少量二氯甲烷溶解产品,再用溶液体积12倍量的无水乙醇进行沉降,抽滤,反复进行3次,最后所得共聚物产品在真空干燥箱中45℃减压干燥3h,得不同配比的PLGA成品。实施例5制备本发明所述的阿莫西林颗粒(规格:0.25g)处方:制备方法:将实施例1中制备的阿莫西林过100目筛,辅料过40目筛,备用;按处方重量配比称取阿莫西林及56%处方量的碳酸氢钠、47%处方量的乙交酯丙交酯共聚物(34.7:65.3)和40%处方量的硬脂富马酸钠,混合均匀,干法制粒,制成细颗粒;加入处方量的羧甲基淀粉钠和剩余处方量的碳酸氢钠及乙交酯丙交酯共聚物(34.7:65.3),混合均匀,干法制粒,制成颗粒;将上述颗粒再与剩余处方量的硬脂富马酸钠、处方内其他辅料混匀后,装袋,即得。实施例6制备本发明所述的阿莫西林颗粒(规格:0.25g)处方:制备方法:将实施例2中制备的阿莫西林过100目筛,辅料过40目筛,备用;按处方重量配比称取阿莫西林及56%处方量的碳酸氢钠、47%处方量的乙交酯丙交酯共聚物(34.7:65.3)和40%处方量的硬脂富马酸钠,混合均匀,干法制粒,制成细颗粒;加入处方量的羧甲基淀粉钠和剩余处方量的碳酸氢钠及乙交酯丙交酯共聚物(34.7:65.3),混合均匀,干法制粒,制成颗粒;将上述颗粒再与剩余处方量的硬脂富马酸钠、处方内其他辅料混匀后,装袋,即得。实施例7制备本发明所述的阿莫西林颗粒(规格:0.125g)处方:制备方法:将实施例3中制备的阿莫西林过100目筛,辅料过40目筛,备用;按处方重量配比称取阿莫西林及56%处方量的碳酸氢钠、47%处方量的乙交酯丙交酯共聚物(34.7:65.3)和40%处方量的硬脂富马酸钠,混合均匀,干法制粒,制成细颗粒;加入处方量的羧甲基淀粉钠和剩余处方量的碳酸氢钠及乙交酯丙交酯共聚物(34.7:65.3),混合均匀,干法制粒,制成颗粒;将上述颗粒再与剩余处方量的硬脂富马酸钠、处方内其他辅料混匀后,装袋,即得。本发明所制备的产品,其流动性、稳定性较市售品均有显著提高。试验例1流动性比较本发明对比例A为依照专利CN201610060313.5实施例1中方法制备得到的阿莫西林原料样品。休止角是检验粉体流动性好坏的最简便的方法,休止角越小,说明摩擦力越小,流动性越好。本试验采用注入法(固定漏斗法)测定实施例1、实施例2和实施例3所制备的新晶型的阿莫西林原料样品、对比例A与阿莫西林原料市售品1、市售品2和市售品3的休止角。将待测样品倒入漏斗,使其轻轻地、均匀地落入圆盘中心,形成一个圆锥体,当物料从粉体斜边沿圆盘边缘中自由落下时停止加料,用量角器测定休止角,测定结果见表1。结果显示本发明所制备产品流动性更好,优于对比例A及市售品。表1休止角测定结果样品实施例1实施例2实施例3对比例A市售品1市售品2市售品3休止角16.219.517.820.847.148.650.9试验例2稳定剂辅料筛选以新晶型的阿莫西林作为活性成分,以不同配比的PLGA作为稳定剂辅料,制备阿莫西林颗粒。制备方法:将实施例1中制备的阿莫西林过100目筛,辅料过40目筛,备用;按处方重量配比称取阿莫西林及56%处方量的碳酸氢钠、47%处方量的一定配比的PLGA和40%处方量的硬脂富马酸钠,混合均匀,干法制粒,制成细颗粒;加入处方量的羧甲基淀粉钠和剩余处方量的碳酸氢钠及一定配比的PLGA,混合均匀,干法制粒,制成颗粒;将上述颗粒再与剩余处方量的硬脂富马酸钠、处方内其他辅料混匀后,装袋,即得。然后对制备的阿莫西林颗粒进行3个月的加速试验,加速试验条件为温度40±2℃、相对湿度75±5%,试验结束后进行质量检测分析,溶化性、含量和有关物的检测方法均为药典方法(中国药典2015年版二部),结果见表2。表2稳定剂辅料筛选试验由表2可见,通过加速3个月试验数据比较,稳定剂辅料乙交酯丙交酯共聚物最佳的配比为34.7:65.3,由其所制备的阿莫西林颗粒的稳定性最佳。试验例3原料稳定性考察本发明对比例A为依照专利CN201610060313.5实施例1中方法制备得到的阿莫西林原料样品。本发明实施例1、实施例2和实施例3所制备的新晶型的阿莫西林原料样品与对比例A、阿莫西林原料市售品1、市售品2和市售品3进行质量检测分析,检测方法均为药典方法(中国药典2015年版二部),结果见表3。中国药典2015年版二部中明确记载了阿莫西林原料的质量标准,要求含量不得少于95.0%,酸度pH值为3.5-5.5,有关物单杂不得大于1.0%,有关物总杂不得大于3.0%,阿莫西林聚合物(简称聚合物)不得过0.15%,水分为12.0%-15.0%,炽灼残渣不得过1.0%。表3阿莫西林原料质量检测分析由表3可见:与对比例A、市售品1、市售品2、市售品3相比,本发明实施例1、实施例2和实施例3所制备的新晶型的阿莫西林原料样品含量更高,有关物更低。特别是实施例1含量高达99.99%,而市售品1、市售品2、市售品3的含量均还不到96%,有关物及聚合物数据也有明显差距,各项数据显示实施例1、实施例2和实施例3所制备的新晶型的阿莫西林原料样品质量更好。试验例4制剂稳定性考察本发明对比例1为依照专利CN201010556659.7实施例1中的方法制备得到的阿莫西林颗粒样品。本发明对比例2为依照专利CN201610060313.5实施例1中的方法制备的原料样品再按本发明实施例5中方法制备的阿莫西林颗粒样品。本发明实施例5、实施例6和实施例7所制备的阿莫西林颗粒样品与对比例1、对比例2、阿莫西林颗粒市售品A进行加速试验,加速试验条件为在温度40±2℃、相对湿度75±5%的条件下,放置3个月。溶化性、含量和有关物的检测方法均为药典方法(中国药典2015年版二部),结果见表4。中国药典2015年版二部中明确记载了阿莫西林颗粒的质量标准,要求含量为90.0%-110.0%,有关物单杂不得大于1.0%,有关物总杂不得大于5.0%。表4加速试验由表4可见:与市售品A、对比例1、对比例2相比,本发明实施例5-7所制备的阿莫西林颗粒样品含量更高,有关物更低。通过3个月加速试验后,实施例5-7所制备的阿莫西林颗粒样品含量降低得更慢,含量均在103%以上,有关物增加得更慢,有关物单杂均在0.30%以下,有关物总杂均在3.0%以下,溶化性项检查均合格;而对比例1和对比例2也符合质量标准,但含量下降和有关物增加得都很快,含量分别为92.6%和96.1%,有关物单杂分别为0.77%和0.59%,有关物总杂分别为4.87%和3.99%,溶化性项检查均合格;而市售品A在加速2个月时,就已出现质量不合格的情况,含量为81.4%,有关物单杂为2.16%,有关物总杂为9.15%,溶化性项检查也不合格。通过3个月加速试验后,市售品A的有关物单杂约为实施例5-7所制备的阿莫西林颗粒样品的25倍,而有关物总杂约为9倍,含量还不到一半,以上结果显示,本发明实施例5-7所制备的阿莫西林颗粒样品的稳定性得到急剧提升。由此可见,本发明提供的阿莫西林颗粒产品,由于使用了本发明制备的特定的晶型形式的阿莫西林并且处方中使用了乙交酯丙交酯共聚物(34.7:65.3),且乙交酯丙交酯共聚物(34.7:65.3)内加与外加重量比为47:53,使得本发明所述的阿莫西林颗粒有关物含量更低,有效成分含量更高,且制剂稳定性大大提高,具有显而易见的效果,为临床应用提供了更好的选择。以上仅是本发明的优选实施方式,并不用以限制本发明,对于本领域技术人员来说,在不脱离本发明原理的前提下,还可以做出的若干改进、润饰、等同替换,均应包含在本发明的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1