抑制多种病毒感染的药物及其联用的制作方法
本发明涉及生物医药领域,具体地,涉及抑制多种病毒感染的药物及其联用。
背景技术
寨卡病毒属黄病毒科、黄病毒属,单股正链rna病毒,是一种通过蚊虫进行传播的虫媒病毒,也可以通过性接触传播、母婴传播。此外它的传播媒介——伊蚊,也是黄病毒科中的另外三种病毒——登革热、基肯孔雅病毒和黄热病毒的媒介。寨卡病毒首次于1947年从黄热病监测的恒河猴体内分离得到。后又于1952年在乌干达及坦桑尼亚发现人感染寨卡病毒。之后的报道都是关于此病毒的小规模流行,直至2007年,在南太平洋地区密克罗尼西亚(micronesia)联邦雅浦(yap)岛发生寨卡病毒爆发性流行。2013年,波利尼西亚发生寨卡病毒爆发疫情。紧接着巴西在2015年5月发现首例寨卡病毒病例。至今全世界已经有30多个国家报道寨卡感病毒感染病例。
寨卡病毒的潜伏期约为数天至一周。临床表现与登革热和基肯孔雅热类似,为急性发热伴斑丘疹、关节痛或结膜炎。目前对于寨卡病毒尚无有效疫苗用于预防,因其主要通过感染病毒的伊蚊类蚊虫叮咬传播,且无临床特效药,故现主要以防蚊控蚊作为预防手段,由于此方法不能彻底杜绝寨卡病毒的增殖和传播,且目前防蚊控蚊的方法为生物控蚊和化学控蚊,都会造成对人类本身带来损伤的副作用,故针对寨卡病毒临床药物的需求显得尤为迫切。
流感病毒属单股负链rna病毒,正粘病毒科。人流感病毒根据核糖核蛋白和基质蛋白可分为甲、乙、丙三种类型。禽流感根据对鸡的致病力分为:高致病性禽流感h5和h7亚型、中致病性禽流感和低/非致病性禽流感。流感病毒的爆发历史悠久,几乎每年都有不同亚型的病毒在全世界流行,例如1980在美国爆发的a/h7n7型病毒、1997年于香港爆发的a/h5n1型禽流感等。
对于流感病毒的治疗,目前有神经氨酸酶类抑制剂,如奥司他韦(oseltamivir)、扎那米韦(zanamivir)、帕拉米韦(peramivir),以及离子通道m2阻滞剂。市场上也已有全部病毒灭活疫苗,基因工程疫苗以及减毒活疫苗出现。然而由于流感病毒的高变异型、高致病性以及流传迅速而使每年都有极大部分人感染。且上述的药物对流感的治疗效果并不十分理想,故对治疗流感的临床类特效药的需求仍然存在。
ev71病毒为二十面体对称球形结构,无包膜,直径约为24~30nm,为单股正链rna病毒。ev71病毒于1969年首次从加利福尼亚患有中枢神经系统疾病的婴儿患者的粪便标本中分离出来。其后全世界许多国家对ev71的病例都有报道。在1998年台湾地区ev71大爆发,129106个手足口病和红斑疹病例中,405例呈严重的中枢神经系统感染,其中有78例因中大部分是由于枢神经系统感染导致的肺水肿和出血而死亡。而目前对于ev71也缺乏特异、高效的抗病毒药物。单纯疱疹病毒(herpessimplexvirus,hsv)能引起人类多种疾病,如龈口炎(gingivostomatitis)、角膜结膜炎(keratoconjunctivitis)、脑炎(encephalitis)以及生殖系统感染和新生儿的感染。在感染宿主后,常在神经细胞中建立潜伏感染,激活后又会出现无症状的排毒,在人群中维持传播链,周而复始地循环。hsv有二个血清型,即hsv-1和hsv-2,hsv-1主要由口唇病灶获得,hsv-2可从生殖器病灶分离到。感染是由于人与人的接触。从发生后四个月到数年被感染的人数可达人口总数的50—90%,是最易侵犯人的一种病毒,但在临床仅有一部份发病。此病可分为:口唇性疱疹、疱疹性角膜炎、疱疹性皮肤炎、阴部疱疹、卡波西病等,有时也是脑膜炎、脑炎的病因。口唇部疱疹一般较易诊断,同时因日晒、发热等种种的刺激因素而引起复发。
新生儿疱疹是临床上常见而又严重的感染,据统计死亡率超过50%,存活者约有1/2严重损伤。hsv-1、hsv-2在分娩时均可通过产道感染新生儿。感染类型有:①皮肤、眼和口腔的局部损伤;②脑炎;③病毒播散到内脏,发生脓毒血症(sepsis),常引起死亡。早期抗感染可减少死亡率。剖腹产是避免生殖道感染的有效方法。妊娠妇女感染hsv-1,病毒有可能经胎盘感染胎儿,造成流产、死胎或先天性畸形。
目前,对疱疹病毒感染的控制尚无特异性有效措施,也没有效果好的疫苗产品,目前的药物也不能彻底防止潜伏感染的再发。因此,研发高效的抗hsv药物是必要的。
因此,本领域迫切需要开发可同时抑制多种病毒感染的药物。
技术实现要素:
本发明的目的在于提供能够抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)增殖的药物。
本发明第一方面提供了一种式i化合物、其水合物、或其药学上可接受盐的用途,用于制备组合物或制剂,所述组合物或制剂用于:
(i)抑制黄病毒科病毒的增殖;和/或
(ii)抑制正粘病毒科病毒的增殖;和/或
(iii)抑制微小病毒科病毒的增殖;和/或
(iv)抑制疱疹病毒的增殖;和/或
(v)治疗黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒的感染;和/或
(vi)上调ifn-β在细胞内的表达;
式中,a为取代或未取代的2-3元杂芳环,所述杂芳环含有1-3个选自n、o或s的杂原子,其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c6烷氧基、c1-c6烷基、c1-c6卤代烷基、c2-c6链烯基、c2-c6链炔基、取代或未取代的噻唑基;
b为取代或未取代的
其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c6烷基、c1-c6卤代烷基、c2-c6链烯基、c2-c6链炔基、取代或未取代的吡嗪酰胺。
在另一优选例中,所述a为取代或未取代的2-3元杂芳环,所述杂芳环含有1-2个选自n、o或s的杂原子,其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c4烷氧基、c1-c4烷基、c1-c4卤代烷基、c2-c4链烯基、c2-c4链炔基、取代或未取代的噻唑基。
在另一优选例中,所述a为取代或未取代的2-3元杂芳环,所述杂芳环含有1个选自n、o或s的杂原子,其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c4烷氧基、c1-c4烷基、c1-c4卤代烷基、c2-c4链烯基、c2-c4链炔基、取代或未取代的噻唑基。
在另一优选例中,所述a为
在另一优选例中,所述b为取代或未取代的
其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c3烷基、c1-c3卤代烷基、c2-c3链烯基、c2-c3链炔基、取代或未取代的吡嗪酰胺。
在另一优选例中,所述b为
在另一优选例中,所述卤素选自下组:f、cl、br或i。
在另一优选例中,所述式i化合物为
在另一优选例中,所述黄病毒科病毒选自下组:寨卡病毒、乙型脑炎病毒、森林脑炎病毒、登革病毒、或其组合。
在另一优选例中,所述正粘病毒科病毒选自下组:流感病毒pr8毒株、甲型流感病毒、乙型流感病毒、丙型流感病毒、或其组合。
在另一优选例中,所述微小病毒科病毒选自下组:肠道病毒71型、小儿麻痹症病毒(poliovirus脊髓灰质炎病毒)、人类鼻病毒a(humanrhinovirusa)、a型肝炎病毒(hepatitisavirus,hav)、脑心肌炎病毒(encephalomyocarditisvirus)、口蹄疫病毒(foot-and-mouthdiseasevirus)、人类副肠内细胞病变人类孤儿病毒(humanparechovirus人副肠孤病毒)、肠道病毒68型、柯萨奇病毒、或其组合。
在另一优选例中,所述疱疹病毒选自下组:单纯疱疹病毒1型(hsv-1)、单纯疱疹病毒2型、水痘带状疱疹病毒、eb病毒、巨细胞病毒、人类疱疹病毒6型、人类疱疹病毒7型、人类疱疹病毒8型、卡波氏肉瘤病毒、或其组合。
在另一优选例中,所述细胞为病毒感染的细胞。
在另一优选例中,所述病毒选自下组:黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、疱疹病毒、或其组合。
在另一优选例中,所述病毒选自下组:寨卡病毒、流感病毒、肠道病毒71型、单纯疱疹病毒(hsv-1)、或其组合。
在另一优选例中,组合物包括药物组合物。
在另一优选例中,所述的组合物还包括其他抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物。
在另一优选例中,所述其他抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物选自下组:利托那韦(ritonavir)、芦平曲韦(rupintrivir,rpt)、扎那米韦(zanamivir)、溴麦角环肽(bromocriptine)、索非布韦(sofosbuvir)、金刚烷胺(amantadine)、金刚乙胺(rimantadine)、奥司他韦(oseltamivir)、苏拉明(suramin)、利巴韦林(ribavirin)、普拉康纳利(pleconaril)、伊曲康唑(itraconazole)、或其组合。
在另一优选例中,所述的药物组合物含有(i)式i所示的化合物、其水合物、或其药学上可接受的盐;以及(ii)药学上可接受的载体。
在另一优选例中,所述组分(i)占所述药物组合物总重量的0.001-99.9wt%,较佳地0.1-99wt%,更佳地1%-90wt%。
在另一优选例中,所述的组合物或药物包括:口服制剂和非口服制剂。
在另一优选例中,所述的制剂包括:气雾剂、喷雾剂、滴鼻剂、直肠栓剂、直肠保留灌肠液、粉剂、颗粒剂、胶囊剂、注射剂、酊剂、口服液、片剂、和/或含片。
在另一优选例中,所述组合物为口服制剂。
在另一优选例中,所述的组合物(如药物组合物)通过以下方式施用于鸟类和/或哺乳动物:口服,静脉、皮下、肌肉或局部注射,粘膜给药(如滴鼻),气雾吸入,或舌下给药。
在另一优选例中,所述哺乳动物包括人或非人哺乳动物。
在另一优选例中,所述非人哺乳动物包括啮齿动物,如小鼠、大鼠、兔;灵长目动物,如猴、猩猩;偶蹄目家畜,如猪、牛、羊;奇蹄目家畜,如马、驴、骡;猫科动物,如家猫;犬科动物,如家犬。
在另一优选例中,所述鸟类包括鸡、鸭、鹅、和/或鸽。
本发明第二方面提供了一种药物组合物,包括:
(a1)第一活性成分,所述第一活性成分为式i化合物、其水合物、或其药学上可接受的盐;
(a2)第二活性成分,所述第二活性成分为抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物;和
(b)药学上可接受的载体;
其中式i化合物的定义如本发明第一方面所述。
在另一优选例中,所述抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物选自下组:利托那韦(ritonavir)、芦平曲韦(rupintrivir,rpt)、扎那米韦(zanamivir)、溴麦角环肽(bromocriptine)、索非布韦(sofosbuvir)、金刚烷胺(amantadine)、金刚乙胺(rimantadine)、奥司他韦(oseltamivir)、苏拉明(suramin)、利巴韦林(ribavirin)、普拉康纳利(pleconaril)、伊曲康唑(itraconazole)、或其组合。
在另一优选例中,所述第一活性成分和第二活性成分的重量比为0.1%-99%,较佳地,20%-80%。
在另一优选例中,所述的药物剂型为口服给药或非口服给药剂型。
在另一优选例中,所述的口服给药剂型是片剂、散剂、颗粒剂或胶囊剂,或乳剂或糖浆剂。
在另一优选例中,所述的非口服给药剂型是注射剂或针剂。
在另一优选例中,所述的式i化合物或其药学上可接受的盐的浓度为0.001μm-50μm,较佳地,0.05μm-20μm,更佳地,0.8μm-10μm。
本发明第三方面提供了一种体外非治疗性的抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的方法,包括步骤:向需要的对象施用如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐,或本发明第二方面所述的药物组合物。
在另一优选例中,所述对象包括人或非人哺乳动物。
在另一优选例中,所述对象包括鸟类。
在另一优选例中,所述非人哺乳动物包括啮齿动物和灵长目动物,优选小鼠、大鼠、兔、猴。
在另一优选例中,所述非人哺乳动物还包括偶蹄目家畜,如猪、牛、羊;奇蹄目家畜,如马、驴、骡;猫科动物,如家猫;犬科动物,如家犬。
在另一优选例中,所述鸟类包括鸡、鸭、鹅、和/或鸽。
在另一优选例中,所述施用剂量为0.1-10mg/kg/天,较佳地,1.0-5mg/kg/天,更佳地,2.0-3mg/kg/天。
在另一优选例中,施用频率为1-6次/天,较佳地1-3次/天。
在另一优选例中,施用包括一个或多个周期,各周期为1-300天,较佳地7-50天。
本发明第四方面提供了一种制备用于抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物组合物的方法,包括步骤:将如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐与药学上可接受的载体混合,从而形成所述药物组合物。
本发明第五方面提供了一种药盒,所述的药盒含有:
(a1)第一容器,以及位于所述第一容器中的如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐,或含有如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐的药物;和
(b1)第二容器,以及位于所述第二容器中的抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物。
在另一优选例中,所述的第一容器和第二容器是相同或不同的容器。
在另一优选例中,所述的第一容器的药物是含如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐的单方制剂。
在另一优选例中,所述的第二容器的药物是含抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的药物的单方制剂。
在另一优选例中,所述药物的剂型为口服剂型或注射剂型。
在另一优选例中,所述药物的剂型包括气雾剂、喷雾剂、滴鼻剂、胶囊、片剂、栓剂、和/或静脉注射剂。
在另一优选例中,所述的试剂盒还含有说明书。
在另一优选例中,所述说明书记载了如下使用方法:
(i)给需要的对象同时施用含有如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐的制剂和含有抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的药物的制剂;和任选的
(ii)重复步骤(i)-(ii)。
本发明第六方面提供了一种体外非治疗性的抑制黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒增殖的方法,包括步骤:
在如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐、或本发明第二方面所述的药物组合物的存在下,培养被所述寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)感染的细胞,从而抑制所述寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)。
本发明第七方面提供了一种治疗黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒感染的方法,包括步骤:
向需要的对象施用含有如本发明第一方面中定义的式i化合物、其水合物、或其药学上可接受的盐的制剂或本发明第二方面所述的药物组合物,从而治疗黄病毒科病毒、正粘病毒科病毒、微小病毒科病毒、和/或疱疹病毒感染。
在另一优选例中,所述对象包括人。
在另一优选例中,所述施用剂量为0.1-10mg/kg/天,较佳地,1.0-5.0mg/kg/天,更佳地,2.0-3.0mg/kg/天。
在另一优选例中,施用频率为1-6次/天,较佳地1-3次/天。
在另一优选例中,施用包括一个或多个周期,各周期为1-300天,较佳地7-50天。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
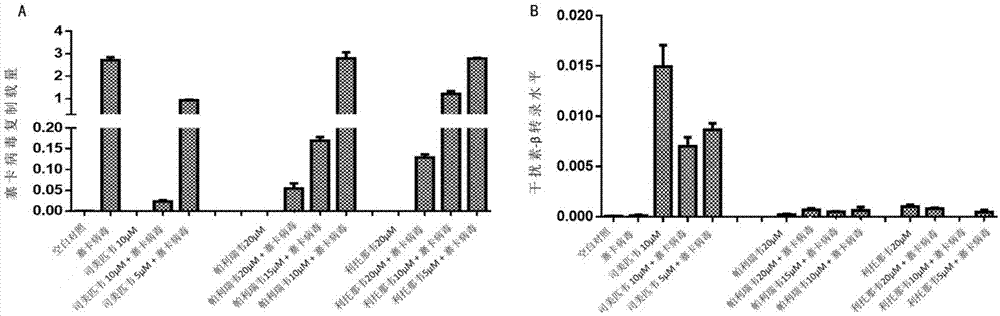
图1显示了不同浓度的司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对zikv病毒的抑制作用。其中,图1,a-b中,将司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)分别用不同的浓度(simeprevir:10um、5um;paritaprevir:20um、15um、10um;ritonavir:20um、10um、5um)加入到vero细胞中,30分钟后,加入zikv病毒(moi=0.1)。72h后收集病毒抽提rna,用real-timepcr检测胞内zikv病毒rna载量(a)以及ifn-β转录水平(b)。
图2显示了不同浓度的司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对zikv病毒的抑制作用。其中,图2,a-b中,将司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)分别用不同的浓度(simeprevir:10um、5um;paritaprevir:20um、15um、10um;ritonavir:20um、10um、5um)加入到u251细胞中,30分钟后,加入zikv病毒(moi=0.1)。72h后收集细胞并抽提rna,用real-timepcr检测胞内zikv病毒rna载量(a)以及ifn-β转录水平(b)。
图3显示了不同浓度的司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对pr8病毒的抑制作用。其中,图3,a-b中,将司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)分别加入到a549细胞中(司美匹韦:10um;帕利瑞韦:40um、20um、1um;利托那韦:10um、5um、1um),30分钟后,加入pr8毒株(moi=1),18小时后收集细胞并抽提rna,用real-timepcr检测胞内pr8病毒rna载量(a)以及ifn-β转录水平(b)。
图4显示了不同浓度的司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对ev71病毒的抑制作用。其中,图4,a-b中,将司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)分别用不同的浓度(司美匹韦:10um;帕利瑞韦:40um、20um、1um;利托那韦:10um、5um、1um)加入到rd细胞中,30分钟后,加入ev71病毒(moi=0.1)。18小时后收集细胞并抽提rna,用real-timepcr检测胞内ev71病毒rna载量(a)以及ifn-β转录水平(b)。
图5显示了不同浓度的司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)对单纯疱症病毒1型(hsv-1)的抑制作用。在图5中,将司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)分别用不同的浓度(司美匹韦:5um、1um、0.2um;帕利瑞韦:20um、10um、5um)加入到vero细胞中,30分钟后,加入hsv-1病毒(moi=10)。24h后收集病毒抽提rna,用real-timepcr检测胞内hsv-1病毒rna载量。
图6显示了司美匹韦(simeprevir,10μm)和帕利瑞韦(paritaprevir,20μm)两种药物分别在寨卡病毒感染(moi=0.1)后的不同时间点处理被感染的非洲绿猴肾细胞系(vero细胞)。寨卡病毒感染72小时后收集细胞并抽提rna,通过realtimepcr检测病毒的复制量。结果显示早期加入药物(感染后12小时)的实验组寨卡病毒复制量很低,此后加入药物的实验组(感染后14小时、16小时、18小时等)寨卡病毒复制依次升高(图6a、6b)。
图7显示了司美匹韦(simeprevir,10μm)和帕利瑞韦(paritaprevir,20μm)两种药物分别在流感病毒pr8株感染(moi=1)后的不同时间点处理被感染的a549细胞。流感病毒pr8株感染24小时后收集细胞并抽提rna,实时定量pcr检测流感病毒的复制量。结果显示早期加入药物(感染后4小时)的实验组流感病毒的复制量最低,此后加入药物的实验组(感染后6小时、8小时等)流感病毒的复制依次升高(图7a、7b)。
图8显示了司美匹韦(simeprevir,10μm)和帕利瑞韦(paritaprevir,20μm)两种药物分别在肠道病毒71型感染后的不同时间点处理被感染的人横纹肌肉瘤细胞(rd细胞)。肠道病毒71型感染24小时后收集细胞并抽提rna,实时定量pcr检测病毒的复制量。结果显示早期加入药物(感染后4小时)的实验组肠道病毒复制量最低,此后加入药物的实验组(感染后6小时、8小时、12小时等)肠道病毒71型复制依次升高(图8a、8b)。
图9显示了司美匹韦(simeprevir,5μm)和帕利瑞韦(paritaprevir,20μm)两种药物分别在单纯疱疹病毒i型感染后的不同时间点处理被感染的vero细胞。单纯疱疹病毒i型感染24小时后裂解细胞并抽提dna,实时定量pcr检测病毒的复制量。结果显示早期加入药物(感染后6小时)的实验组单纯疱疹病毒i型复制量最低,此后加入药物的实验组(感染后12小时、15小时、18小时等)单纯疱疹病毒i型复制依次升高(图9a、9b)。
图10显示了司美匹韦(simeprevir,2μm)、帕利瑞韦(paritaprevir,15μm)、溴麦角环肽(2μm)以及前两者分别和溴麦角环肽联用处理寨卡病毒感染的vero细胞。寨卡病毒感染72小时后收集细胞并抽提rna,实时定量pcr检测寨卡病毒的复制量。结果显示司美匹韦、溴麦角环肽单独处理的实验组寨卡病毒复制较加二甲基亚砜(dmso)对照组低;两者联合使用显著降低寨卡病毒的复制(图10a)。此外,帕利瑞韦单独处理实验组寨卡病毒复制也较加二甲基亚砜(dmso)对照组低,帕利瑞韦和溴麦角环肽联合使用时也显著降低寨卡病毒的复制(图10b)。
图11显示了司美匹韦(simeprevir,2μm)、帕利瑞韦(paritaprevir,15μm)、伊曲康唑(0.2μm)以及前两者分别和伊曲康唑联用处理肠道病毒71型感染的rd细胞。肠道病毒71型感染24小时后收集细胞并抽提rna,实时定量pcr检测肠道病毒71型的复制量。结果显示司美匹韦、伊曲康唑单独处理的实验组肠道病毒71型复制较加二甲基亚砜(dmso)对照组低;两者联合使用显著降低肠道病毒71型的复制(图11a)。此外,帕利瑞韦单独处理组肠道病毒71型复制也较加二甲基亚砜(dmso)的对照组低,帕利瑞韦和伊曲康唑联合使用也显著降低肠道病毒71型的复制(图11b)。
具体实施方式
本发明人通过广泛而深入的研究,通过筛选大量的化合物,首次意外发现,式i化合物、其水合物、或其药学上可接受的盐具有抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)增殖的作用,此外,本发明还首次发现,将式i化合物、其水合物、或其药学上可接受的盐与其他抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)增殖的药物(如利托那韦(ritonavir)、芦平曲韦(rupintrivir,rpt)、扎那米韦(zanamivir)、溴麦角环肽(bromocriptine)、索非布韦(sofosbuvir)、金刚烷胺(amantadine)、金刚乙胺(rimantadine)、奥司他韦(oseltamivir)、苏拉明(suramin)、利巴韦林(ribavirin)、普拉康纳利(pleconaril)等)联用,可更有效地抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)的增殖,从而治疗黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)的感染。在此基础上,本发明人完成了本发明。
基团定义
如本文所用,术语“取代或未取代的”指所述基团可以是未取代的,或者所述基团中的h被一个或多个(如1-10个,较佳地1-5个,更佳地1-3个,最佳地,1-2个)取代基所取代。
如本文所用,所述的“取代”或“取代的”指所述基团具有一个或多个(较佳地1-6个,更佳地1-3个)选自下组的取代基:卤素、c1-c6烷基、c1-c6卤代烷基、c2-c6链烯基、c2-c6链炔基。
如本文所用,术语“c1-c6烷基”是指具有1-6个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、或类似基团。
如本文所用,术语“c2-c6链烯基”指具有2-6个碳原子的直链或支链的烯基,例如乙烯基、烯丙基、1-丙烯基、异丙烯基、1-丁烯基、2-丁烯基、或类似基团。
如本文所用,术语“c2-c6链炔基”是指具有2-6个碳原子的直链或支链的炔基,例如乙炔基、丙炔基、或类似基团。
如本文所用,术语“c1-c6卤代烷基”是指氢被1个或1个以上的卤素取代的具有1-6个碳原子的直链或支链烷基,例如,卤代甲基、卤代乙基、卤代丙基、卤代异丙基、或类似基团。
如本文所用,术语“c1-c6烷氧基”是指具有(c1-c6烷基)-o-结构的基团,例如,ch3-o-、c2h5-o-、c3h8-o-、或类似基团。
如本文所用,术语“c1-c4烷基”是指具有1-4个碳原子的直链或支链烷基,例如甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、或类似基团。
如本文所用,术语“c2-c4链烯基”指具有2-4个碳原子的直链或支链的烯基,例如乙烯基、烯丙基、1-丙烯基、异丙烯基、1-丁烯基、2-丁烯基、或类似基团。
如本文所用,术语“c2-c4链炔基”是指具有2-4个碳原子的直链或支链的炔基,例如乙炔基、丙炔基、或类似基团。
如本文所用,术语“c1-c4卤代烷基”是指氢被1个或1个以上的卤素取代的具有1-4个碳原子的直链或支链烷基,例如,卤代甲基、卤代乙基、卤代丙基、卤代异丙基、或类似基团。
如本文所用,术语“c1-c4烷氧基”是指具有(c1-c4烷基)-o-结构的基团,例如,ch3-o-、c2h5-o-、c3h7-o-、或类似基团。
如本文所用,术语“卤素”是指氟、氯、溴、或碘,优选氟和氯。
如本文所用,术语“卤代的”指被相同或不同的一个或多个上述卤原子取代的基团,可以部分卤代或全部卤代,例如三氟甲基、五氟乙基、七氟异丙基、或类似基团。
本发明的化合物可以含有一个或多个不对称中心,并因此以消旋体、外消旋混合物、单一对映体、非对映异构体化合物和单一非对映体的形式出现。可以存在的不对称中心,取决于分子上各种取代基的性质。每个这种不对称中心将独立地产生两个旋光异构体,并且所有可能的旋光异构体和非对映体混合物和纯或部分纯的化合物包括在本发明的范围之内。本发明包括化合物的所有异构形式。
活性成分
如本文所用,所述“本发明的活性成分”、“式i化合物”可互换使用,均指能够有效抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)的增殖的化合物,
式中,a为取代或未取代的2-3元杂芳环,所述杂芳环含有1-3个选自n、o或s的杂原子,其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c6烷氧基、c1-c6烷基、c1-c6卤代烷基、c2-c6链烯基、c2-c6链炔基、取代或未取代的噻唑基;
b为取代或未取代的
其中,所述“取代的”是指基团中的h被选自下组的一个或多个取代基所取代:卤素、c1-c6烷基、c1-c6卤代烷基、c2-c6链烯基、c2-c6链炔基、取代或未取代的吡嗪酰胺。
在一优选实施方式中,所述化合物具有式ia或式ib所示的结构:
一类特别优选的式i化合物为实施例所述的化合物,即simeprevir和paritaprevir,结构式分别为
在本发明中,还包括式i化合物的药学上可接受的盐。术语“药学上可接受的盐”指本发明化合物与酸或碱所形成的适合用作药物的盐。药学上可接受的盐包括无机盐和有机盐。一类优选的盐是本发明化合物与酸形成的盐。适合形成盐的酸包括但并不限于:盐酸、氢溴酸、氢氟酸、硫酸、硝酸、磷酸等无机酸,甲酸、乙酸、丙酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、乳酸、苹果酸、酒石酸、柠檬酸、苦味酸、甲磺酸、苯甲磺酸,苯磺酸等有机酸;以及天冬氨酸、谷氨酸等酸性氨基酸。
组合物和施用方法
如本文所用,术语“组合物”包括(a1)第一活性成分,所述第一活性成分为式i化合物、其水合物或其药学上可接受的盐;和(a2)第二活性成分,所述第二活性成分为抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的药物;和(b)药学上可接受的载体;其中式i化合物的定义如本发明第一方面中所述。此外,所述的组合物包括药物组合物、食品组合物或保健品组合物。
通常,可将本发明的活性成分配制于无毒的、惰性的和药学上可接受的载体介质。配制好的药物组合物可以通过常规途径进行给药,其中包括(但并不限于):口服、肌内、腹膜内、静脉内、皮下、皮内、或局部给药。
本发明还提供了一种药物组合物,它含有安全有效量的本发明的活性成分以及药学上可接受的载体或赋形剂。这类载体包括(但并不限于):盐水、缓冲液、葡萄糖、水、甘油、乙醇、及其组合。药物制剂应与给药方式相匹配。本发明的药物组合物可以被制成针剂形式,例如用生理盐水或含有葡萄糖和其他辅剂的水溶液通过常规方法进行制备。诸如片剂和胶囊之类的药物组合物,可通过常规方法进行制备。药物组合物如针剂、溶液、片剂和胶囊宜在无菌条件下制造。活性成分的给药量是治疗有效量,例如每天约1微克-10毫克/千克体重,优选地,甘草利酮或其衍生物的用量可以为:成年人每日0.1~2000mg,优选1~300mg/天。
作为抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)的感染,可以制成口服和非口服制剂。口服给药可制成片剂、散剂、颗粒剂、胶囊剂等常用剂型,所用的赋型剂可以为淀粉、乳糖、蔗糖、甘露糖、羟甲基纤维素等中的一种或几种。崩解剂可以为马铃薯淀粉、羟甲基纤维素等中的一种或几种。粘合剂可以为阿拉伯胶、玉米淀粉、明胶、糊精等中的一种或几种。口服制剂除上述剂型外,还可以制成乳剂、糖浆剂等。
非口服制剂可以制成注射剂,可以与注射用水、生理盐水、葡萄糖水制成注射剂,也可以在其中加入一定比例的乙醇、丙醇、乙二醇等。此外也可制成滴鼻剂、喷雾剂、直肠栓剂、直肠保留灌肠液等常用剂型。
此外,本发明的活性成分还特别适合与其他抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)增殖的药物联用。尤其是本发明的活性成分可与利托那韦(ritonavir)、芦平曲韦(rupintrivir,rpt)、扎那米韦(zanamivir)、溴麦角环肽(bromocriptine)、索非布韦(sofosbuvir)、金刚烷胺(amantadine)、金刚乙胺(rimantadine)、奥司他韦(oseltamivir)、苏拉明(suramin)、利巴韦林(ribavirin)、普拉康纳利(pleconaril)等联合使用,从而更有效的抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)的感染。
本发明的进一步目的是提供一种抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的制备方法,采用所述的式i所示的化合物及其衍生物为药物原料,用相应的赋型剂,按照常规的方法制成口服和非口服制剂,其中式i所示的化合物及其衍生物的用量可以为:成年人每日0.1~2000mg,优选1~300mg/天,每日服用1~5次;儿童的用量和次数需在成人的基础上酌情递减。
药盒
本发明还提供了一种药盒(或本发明第一方面所述抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的药物),所述的药盒含有:
(a1)第一容器,以及位于所述第一容器中的如权利要求1中定义的式i化合物或其药学上可接受的盐,或含有如权利要求1中定义的式i化合物、其水合物、或其药学上可接受的盐的药物;和
(b1)第二容器,以及位于所述第二容器中的抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的药物。
在一优选实施方式中,所述的第一容器和第二容器是相同或不同的容器。
所述含有式i所示的化合物及其衍生物的制剂可以是含有式i化合物及其衍生物的单元剂型,所述含有抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的制剂可以是含有抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)增殖药物的单元剂型。
如本文所用,术语“单元剂型”是指为了服用方便,将组合物制备成单次服用所需的剂型,包括但不限于各种固体剂(如片剂)、液体剂、胶囊剂、缓释剂。
在另一优选例中,所述说明书记载了如下使用方法:
(i)给需要的对象同时施用含有如权利要求1中定义的式i化合物、其水合物、或其药学上可接受的盐的制剂和含有抑制黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)增殖的药物的制剂;和任选的
(ii)重复步骤(i)-(ii)。
本发明制剂可以每一天服用三次到每十天服用一次,或者以缓释方式每十天服用一次。优选的方式是每天服用一次,因为这样便于病人坚持,从而显著提高病人服药的顺应性。
服用时,极大多数病例一般每天应用的总剂量应低于(或少数病例等于或略大于)各个单药的每天常用剂量,当然,所用的活性成分的有效剂量可随给药的模式和待治疗的疾病的严重程度等而有所变化。
本发明的主要优点包括:
(1)本发明通过大量筛选,首次发现式i化合物具有抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的活性。
(2)本发明首次发现式i化合物可与其他抑制寨卡病毒、流感病毒、肠道病毒71型、和/或单纯疱疹病毒(hsv-1)增殖的药物联用,从而更有效的治疗黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)的感染。
(3)本发明首次发现,式i化合物可作为活性成分,从而制备治疗黄病毒科病毒(如寨卡病毒)、正粘病毒科病毒(如流感病毒pr8毒株)、微小病毒科病毒(如肠道病毒71型)、和/或疱疹病毒(如单纯疱疹病毒1型,hsv-1)感染的药物。
(4)本发明为临床上对于由微小病毒科病毒、正粘病毒科病毒、黄病毒科病毒和疱疹病毒所引起的各部分疾病的药物治疗提供了一定的理论基础,具有重要的学术价值和应用价值。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数是重量百分比和重量份数。
如无特别说明,则本发明说明书中的材料和试剂均为市售产品。
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)购自mce中国(上海皓元生物医药科技有限公司)。
利托那韦(ritonavir)、芦平曲韦(rupintrivir,rpt)、扎那米韦(zanamivir)、溴麦角环肽(bromocriptine)、索非布韦(sofosbuvir)、金刚烷胺(amantadine)、金刚乙胺(rimantadine)、奥司他韦(oseltamivir)、苏拉明(suramin)、利巴韦林(ribavirin)、普拉康纳利(pleconaril)购自mce中国(上海皓元生物医药科技有限公司)。
simeprevir为西咪匹韦,结构式为:
paripaprevir为viekirax,中文名帕利瑞韦,结构式为:
ritonavir为利托那韦,结构式为:
芦平曲韦(rupintrivir,rpt),结构式为:
扎那米韦(zanamivir),结构式为:
溴麦角环肽(bromocriptine,多巴胺d2/d3受体激动剂),结构式为:
索非布韦(sofosbuvir),结构式为:
金刚烷胺(amantadine),结构式为:
金刚乙胺(rimantadine),结构式为:
奥司他韦(oseltamivir),结构式为:
苏拉明(suramin),结构式为:
利巴韦林(ribavirin),结构式为:
普拉康纳利(pleconaril),结构式为:
伊曲康唑(itraconazole),结构式为
实施例1
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对寨卡病毒(zikv)的抑制作用
将司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)这三种药物以不同浓度分别加入到非洲绿猴肾细胞系(vero)(购自美国模式培养物集存库(americantypeculturecollection,atcc)中,然后加入zikv病毒(moi=0.1)(获自中科院上海巴斯德研究所),再收集细胞并抽提rna,通过real-timepcr方法检测细胞中病毒rna载量,发现加了上述药物的细胞中病毒载量降低,并呈浓度依赖关系(图1a),同时发现simeprevir能够上调细胞内ifn-β转录水平(图1b),说明这三种hcv药物能够抑制zikv在其宿主细胞中的增殖,并且司美匹韦可能通过上调细胞ifn-β从而增强细胞的抗病毒能力。
实施例2
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对寨卡病毒(zikv)的抑制作用
在另外一组以zikv感染的人胶质瘤细胞系(u251)(购自美国模式培养物集存库(americantypeculturecollection,atcc)体系中,同样以实施例1的上述方法检测,发现与实施例1的结果相同,更进一步说明了zikv能够被司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)这三类药所抑制(图2a),并且此实验中司美匹韦同样能上调干扰素-β在胞内的转录水平(图2b)。
实施例3
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对流感病毒的抑制作用
用实施例1相同的方法,发明人发现在上述三种药物也能够抑制流感病毒株pr8在人非小细胞肺癌细胞系(a549细胞)(购自美国模式培养物集存库(americantypeculturecollection,atcc)中的增殖,并呈浓度依赖关系(图3a)。与此同时,pr8毒株本身能够引发胞内干扰素-β的高表达,在用上述药物进行干扰后,胞内干扰素-β的转录水平明显降低(图3b),这说明流感毒株pr8能够明显被这三种药物所抑制。
实施例4
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对ev71病毒的抑制作用
用实施例1相同的方法,以人横纹肌肉瘤细胞(rd)(购自美国模式培养物集存库(americantypeculturecollection,atcc)为宿主细胞,检测了这三种药物对ev71病毒扩增的影响。从实验结果能够清楚的看出,ev71在胞内的复制扩增能够被这三种药物所抑制,并呈浓度梯度依赖关系(图4a)。而单独的ev71本身能够引起细胞内ifn-β转录水平的上调,在加入上述三种药物后,都能达到降低ifn-β转录水平的效果(图4b)。
综上所述,司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)三种hcv药物对zikv(黄病毒科病毒)、流感pr8毒株(正粘病毒科病毒)和ev71病毒(微小病毒科病毒)在胞内的复制和扩增都有很明显的抑制作用,同时单独的司美匹韦能够上调胞内干扰素-β的转录水平。这些现象均为它们在临床上实现对相关病毒所引发的不同疾病的治疗提供了一定的理论基础。
实施例5
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对hsv-1病毒的抑制作用
用实施例1相同的方法,发现上述三种药物也能够抑制hsv-1病毒(疱疹病毒)在非洲绿猴肾细胞系(vero)(购自美国模式培养物集存库(americantypeculturecollection,atcc)中的增殖,并呈浓度依赖关系(图5)。
实施例6
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)对zikv、ev71、pr8和hsv-1病毒的抑制作用
通过上述试验,不同药物(司美匹韦、帕利瑞韦和利托那韦)在各自浓度条件下,对不同病毒(zikv、ev71、pr8和hsv-1)增殖的抑制作用,最后通过real-timepcr的方法,测得病毒被药物抑制后的载量数据,并将此数据与没有加入药物的病毒复制载量的对照组进行对比,得出药物对病毒的抑制效率。
从表1、表2的结果可以看出,司美匹韦对于zikv、ev71和pr8,在10um时能够达到很好的抑制效果;对于zikv,在15um的浓度时抑制效果较好,而对于ev71和pr8,帕利瑞韦则在40um的浓度条件下有着很好的抑制效果;对于zikv,在20um的条件下抑制效果较好,且在10um的条件下,对pr8的抑制效果较好,而对于ev71,利托那韦在10um的条件下抑制率为46.7921,这说明,随着药物浓度的增加,利托那韦对ev71的抑制效果更好。另外,表3显示出,司美匹韦在5um时对hsv-1的抑制效果非常好,达到了99.55%,而帕利瑞韦在20um的浓度条件下对hsv-1的抑制率达到了93.33%。
表1
表2
表3
实施例7司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)联用对zikv、ev71、pr8和hsv-1病毒的抑制作用
在本发明中,用real-timepcr测病毒载量的方法,研究了司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)的联用对zikv、ev71、pr8和hsv-1病毒的抑制效果,结果表明,联用司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)和利托那韦(ritonavir)可更有效的抑制zikv、ev71、pr8和hsv-1病毒,抑制率可分别达到80-99.99%,40-99%,90-99.99%和50-99.99%。
实施例8
司美匹韦(simeprevir)和帕利瑞韦(paritaprevir)抑制寨卡病毒在细胞内的复制过程
司美匹韦(10μm)和帕利瑞韦(20μm)两种药物分别在寨卡病毒感染(moi=0.1)后的不同时间点处理被感染的非洲绿猴肾细胞系(vero细胞,购自美国模式培养物集存库,americantypeculturecollection,atcc)。寨卡病毒感染72小时后,实时定量pcr(realtimepcr)检测病毒的复制量。实验数据显示早期加入药物(感染后12小时加药)的实验组寨卡病毒复制量很低,此后加入药物的实验组(感染后14小时、16小时、18小时等时间点加药)寨卡病毒复制依次升高(图6a、6b)。这说明司美匹韦和帕利瑞韦抑制寨卡病毒的复制过程。
实施例9
司美匹韦(simeprevir)和帕利瑞韦(paritaprevir)抑制流感病毒pr8株在细胞内的复制过程
司美匹韦(10μm)和帕利瑞韦(20μm)两种药物分别在流感病毒pr8株感染(moi=1)后的不同时间点处理被感染的a549细胞(购自美国模式培养物集存库,americantypeculturecollection,atcc)。流感病毒pr8株感染24小时后,实时定量pcr检测病毒的复制量。实验数据显示早期加入药物(感染后4小时)的实验组流感病毒pr8株的复制量最低,此后加入药物的实验组(感染后6小时、8小时等)流感病毒pr8株复制依次升高(图7a、7b)。这说明司美匹韦和帕利瑞韦抑制流感病毒pr8株的复制过程。
实施例10
司美匹韦(simeprevir)和帕利瑞韦(paritaprevir)抑制肠道病毒71型在细胞内的复制过程
司美匹韦(10μm)和帕利瑞韦(20μm)两种药物分别在肠道病毒71型感染后的不同时间点处理被感染的人横纹肌肉瘤细胞(rd细胞,购自美国模式培养物集存库,americantypeculturecollection,atcc)。肠道病毒71型感染24小时后,实时定量pcr检测病毒的复制量。实验数据显示早期加入药物(感染后4小时)的实验组肠道病毒71型复制量最低,此后加入药物的实验组(感染后6小时、8小时、12小时)肠道病毒71型复制依次升高(图8a、8b)。这说明司美匹韦和帕利瑞韦抑制肠道病毒71型的复制过程。
实施例11
司美匹韦(simeprevir)和帕利瑞韦(paritaprevir)抑制单纯疱疹病毒i型在胞内的复制过程
司美匹韦(5μm)和帕利瑞韦(20μm)两种药物分别在单纯疱疹病毒i型感染后的不同时间点处理被感染的vero细胞。单纯疱疹病毒i型感染24小时后裂解细胞并抽提dna,实时定量pcr检测病毒的复制量。实验数据显示早期加入药物(感染后6小时)的实验组单纯疱疹病毒i型复制量最低,此后加入药物的实验组(感染后12小时、15小时、18小时)单纯疱疹病毒i型复制依次升高(图9a、9b)。这说明司美匹韦和帕利瑞韦抑制单纯疱疹病毒i型的复制过程。
实施例12
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)分别和溴麦角环肽联用更好地抑制寨卡病毒的复制
司美匹韦(2μm)、帕利瑞韦(15μm)、溴麦角环肽(2μm)以及前两者分别和溴麦角环肽联用处理寨卡病毒感染的vero细胞。寨卡病毒感染72小时后,实时定量pcr检测寨卡病毒的复制量。实验数据显示司美匹韦、溴麦角环肽单独处理的实验组寨卡病毒复制较加二甲基亚砜(dmso)对照组低;两者联合使用显著降低寨卡病毒的复制(图10a)。此外,帕利瑞韦单独处理实验组寨卡病毒复制也较加二甲基亚砜(dmso)对照组低,帕利瑞韦和溴麦角环肽联合使用时显著降低寨卡病毒的复制(图10b)。这说明司美匹韦、帕利瑞韦分别与溴麦角环肽联用对寨卡病毒的复制都有较好的抑制效果。
实施例13
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)分别和伊曲康唑联用更好地抑制肠道病毒71型的复制
司美匹韦(2μm)、帕利瑞韦(15μm)、伊曲康唑(0.2μm)以及前两者分别和伊曲康唑联用处理肠道病毒71型感染的rd细胞。肠道病毒71型感染24小时后,实时定量pcr检测肠道病毒71型的复制量。实验数据显示司美匹韦、伊曲康唑单独处理的实验组肠道病毒71型复制较加二甲基亚砜(dmso)对照组低;两者联合使用显著降低肠道病毒71型的复制(图11a)。此外,帕利瑞韦单独处理实验组肠道病毒71型复制也较加二甲基亚砜(dmso)的对照组低,帕利瑞韦和伊曲康唑联合使用显著降低肠道病毒71型的复制(图11b)。这说明司美匹韦、帕利瑞韦分别与伊曲康唑联用对肠道病毒71型复制都有较好的抑制效果。
实施例14
司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)分别和溴麦角环肽(bromocriptine)、伊曲康唑(itraconazole)联用对寨卡病毒、肠道病毒71型的抑制作用
在本发明中,用real-timepcr方法检测病毒复制量的方法,研究司美匹韦(simeprevir)、帕利瑞韦(paritaprevir)分别和溴麦角环肽(bromocriptine)、伊曲康唑(itraconazole)联用对zikv、ev71病毒复制的抑制效果。从表4结果可以看出,低剂量的司美匹韦(2μm)和溴麦角环肽(2μm)对寨卡病毒复制的抑制效率分别为57.7335%和63.5818%时,两者联合使用时的抑制效率达到96.1255%;当低剂量的帕利瑞韦(15μm)和溴麦角环肽(2μm)对寨卡病毒复制的抑制效率分别为55.5798%和84.7913%时,两者联合使用的抑制效率达到93.2442%。
同样地,低剂量的司美匹韦(2μm)和伊曲康唑(0.2μm)对肠道病毒71型的抑制效率分别为50.5471%和71.4757%时,两者联合使用的抑制效率为94.7373%;当低剂量的帕利瑞韦(15μm)和伊曲康唑(0.2μm)对肠道病毒71型的抑制效率分别为25.6938%和30.2939%时,两者联合使用的效率为93.3040%。这些数据进一步确认司美匹韦和帕利瑞韦对寨卡病毒和肠道病毒71型复制的抑制作用。
同时提示,与溴麦角环肽联合使用时,使用较低剂量的司美匹韦和帕利瑞韦抑制寨卡病毒复制可得到很好的效果;与伊曲康唑联合使用时,使用较低剂量的司美匹韦和帕利瑞韦抑制肠道病毒71型可得到很好的效果。
表4
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!