一种基于子种群协同进化的蛋白质结构预测方法与流程
本发明涉及一种生物学信息学、智能优化、计算机应用领域,尤其涉及的是,一种基于子种群协同进化的蛋白质结构预测方法。
背景技术:
生物细胞中包含许多由20多种氨基酸所形成的长链折叠而成的蛋白质。dna以三个核苷酸为一组的密码子转译为蛋白质的氨基酸序列,相对于第一遗传密码,蛋白质序列一级结构与其三级结构之间的对应关系(即第二遗传密码或称折叠密码)仍为未解之谜。然而,蛋白质三级结构折叠错误,会导致糖尿病、白内障、老年痴呆等蛋白质折叠病。因此,知道对应的蛋白质三维结构,是实现这些蛋白质折叠病治疗的先决条件。
目前,测定蛋白质三维结构的实验方法主要包括x射线晶体衍射和多维核磁共振(nmr)。x射线晶体衍射是目前测定蛋白质结构最有效的方法,所达到的精度是其它方法所不能比拟的,主要缺点是蛋白质晶体难以培养且晶体结构测定的周期较长;nmr方法可以直接测定蛋白质在溶液中的构象,但是对样品的需要量大、纯度要求高,目前只能测定小分子蛋白质。其次,这些实验测定方法价格昂贵,测定一个蛋白质的三维结构需要几十万美元,然而,测定一个蛋白质的一级氨基酸序列仅需1000美元左右,从而导致蛋白质序列和三维结构测定之间的鸿沟越来越大。因此,如何以计算机为工具,运用适当的算法,从氨基酸序列出发直接预测蛋白质的三维结构,成为当前生物信息学中一种重要的研究课题。
构象空间优化(或称采样)方法是目前制约蛋白质结构从头预测精度最关键的因素之一。进化算法(evolutionalgorithm,ea)是研究蛋白质分子构象优化的一类重要方法,主要包括遗传算法(geneticalgorithms,ga)、差分进化算法(differentialevolution,de)及分布估计算法(estimationofdistributionalgorithm,eda)。de算法自1995年由price和storn提出以来,在蛋白质构象空间优化领域有了广泛的应用。shehu研究小组基于de算法,提出一系列有效的蛋白质构象空间优化方法,如多尺度混合进化算法hea,多目标构象空间优化方法moea,基于数据驱动的蛋白质能量空间映射方法pca-ea等。在de算法的框架下,张贵军课题组提出了基于抽象凸内核空间引导的构象优化方法和基于片段结构谱知识引导的构象优化方法。然而,在上述优化方法中,由于算法的贪婪性,种群多样性较低,从而导致算法陷入局部最优而影响预测精度;其次,后期收敛速度较慢,从而导致构象搜索效率较低。
因此,现有的构象空间优化方法在预测精度和搜索效率方面存在着缺陷,需要改进。
技术实现要素:
为了克服现有蛋白质构象空间优化方法的预测精度和搜索效率较低的不足,本发明提出一种预测精度和搜索效率均较高的基于子种群协同进化的蛋白质结构预测方法。
本发明解决其技术问题所采用的技术方案是:
一种基于子种群协同进化的蛋白质结构预测方法,所述方法包括以下步骤:
1)输入待测蛋白质的序列信息,并从robetta服务器(http://www.robetta.org/)上得到片段库;
2)参数设置:设置种群规模np,子种群数量n,交叉概率cr,片段长度l,温度因子kt,最大迭代次数gmax,并初始化迭代次数g=0;
3)从对各残基位对应的片段库中随机选择片段组装生成初始构象种群p={c1,c2,...,cnp},并将整个种群随机分成n个大小相等的子种群,其中,n≥4,ci,i={1,2,…,np}为种群p中的第i个构象个体;
4)对种群中的每个构象ci,i∈{1,2,…,np}作如下处理:
4.1)如果构象ci属于第一个子种群,则进行如下操作:
4.1.1)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体ca;
4.1.2)从其余n-2个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cb;
4.1.3)分别从构象个体ca和cb中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.2)如果构象ci属于第二个子种群,则进行如下操作:
4.2.1)根据rosettascore3能量函数计算构象ci所属的子种群的中的每个构象的能量,并选择能量最低的构象记作clbest;
4.2.2)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cc;
4.2.3)分别从构象个体clbest和cc中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.3)如果构象ci属于第三个子种群,则进行如下操作:
4.3.1)根据rosettascore3能量函数计算整个种群中每个构象个体的能量,并按照能量从低到高进行排序;
4.3.2)从排名靠前的np/5种群中随机选择一个构象个体记作cpbest;
4.3.3)从构象ci所属的子种群中随机选择一个与ci和cpbest均不相同的构象个体cd;
4.3.4)分别从构象个体cpbest和cd中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.4)如果构象ci属于其余n-3个子种群,则进行如下操作:
4.4.1)从构象ci所属的子种群中随机选择一个与ci不同的构象ce;
4.4.2)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cf;
4.4.3)分别从构象个体ce和cf中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.5)随机生成一个0和1之间的小数r,如果r<cr,则从构象ci中随机选取一个长度为l的片段替换变异构象cmutant中对应位置的片段,并进行一次随机片段组装,从而生成测试构象ctrial,否则直接将变异构象进行一次随机片段组装生成测试构象ctrial;
4.6)根据rosettascore3能量函数分别计算测试构象ctrial和ci的能量值,如果ctrial的能量值小于ci的能量值,则ctrial替换ci,否则根据玻尔兹曼概率
5)g=g+1,如果g>gmax,则输出能量最低的构象作为最终预测结构,否则返回步骤4)。
本发明的技术构思为:首先,将整个种群划分成多个规模相等的子种群;然后,针对第一个子种群,从其余不同的子种群中选择构象进行变异;针对第二个子种群,选择目标构象所在的子种群中的最优构象以及其余子种群中的构象进行变异;针对第三个子种群,选择整个种群中的最优构象以及其余子种群中的构象进行变异;针对其余子种群,从目标构象所在的子种群中以及其余子种群中选择构象进行变异;最后,根据玻尔兹曼概率对测试构象进行更新。本发明提供一种预测精度和搜索效率均较高的基于子种群协同进化的蛋白质结构预测方法。
本发明的有益效果表现在:针对不同的子种群,通过各子种群中的协同合作进行变异策略,提高种群的多样性,避免陷入局部最优,提高预测精度,同时提高搜索效率。
附图说明
图1是基于子种群协同进化的蛋白质结构预测方法对蛋白质2mqk进行结构预测时的构象更新示意图。
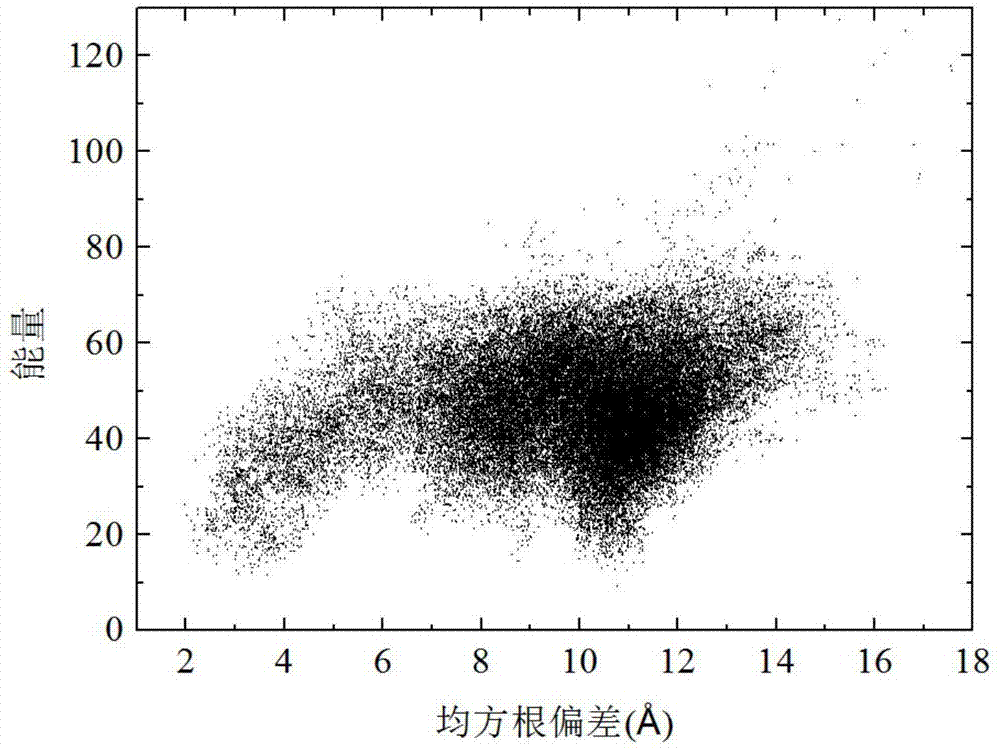
图2是基于子种群协同进化的蛋白质结构预测方法对蛋白质2mqk进行结构预测时得到的构象分布图。
图3是基于子种群协同进化的蛋白质结构预测方法对蛋白质2mqk进行结构预测得到的三维结构图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1~图3,一种基于子种群协同进化的蛋白质结构预测方法,包括以下步骤:
1)输入待测蛋白质的序列信息,并从robetta服务器(http://www.robetta.org/)上得到片段库;
2)参数设置:设置种群规模np,子种群数量n,交叉概率cr,片段长度l,温度因子kt,最大迭代次数gmax,并初始化迭代次数g=0;
3)从对各残基位对应的片段库中随机选择片段组装生成初始构象种群p={c1,c2,...,cnp},并将整个种群随机分成n个大小相等的子种群,其中,n≥4,ci,i={1,2,…,np}为种群p中的第i个构象个体;
4)对种群中的每个构象ci,i∈{1,2,…,np}作如下处理:
4.1)如果构象ci属于第一个子种群,则进行如下操作:
4.1.1)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体ca;
4.1.2)从其余n-2个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cb;
4.1.3)分别从构象个体ca和cb中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.2)如果构象ci属于第二个子种群,则进行如下操作:
4.2.1)根据rosettascore3能量函数计算构象ci所属的子种群的中的每个构象的能量,并选择能量最低的构象记作clbest;
4.2.2)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cc;
4.2.3)分别从构象个体clbest和cc中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.3)如果构象ci属于第三个子种群,则进行如下操作:
4.3.1)根据rosettascore3能量函数计算整个种群中每个构象个体的能量,并按照能量从低到高进行排序;
4.3.2)从排名靠前的np/5种群中随机选择一个构象个体记作cpbest;
4.3.3)从构象ci所属的子种群中随机选择一个与ci和cpbest均不相同的构象个体cd;
4.3.4)分别从构象个体cpbest和cd中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.4)如果构象ci属于其余n-3个子种群,则进行如下操作:
4.4.1)从构象ci所属的子种群中随机选择一个与ci不同的构象ce;
4.4.2)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cf;
4.4.3)分别从构象个体ce和cf中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.5)随机生成一个0和1之间的小数r,如果r<cr,则从构象ci中随机选取一个长度为l的片段替换变异构象cmutant中对应位置的片段,并进行一次随机片段组装,从而生成测试构象ctrial,否则直接将变异构象进行一次随机片段组装生成测试构象ctrial;
4.6)根据rosettascore3能量函数分别计算测试构象ctrial和ci的能量值,如果ctrial的能量值小于ci的能量值,则ctrial替换ci,否则根据玻尔兹曼概率
5)g=g+1,如果g>gmax,则输出能量最低的构象作为最终预测结构,否则返回步骤4)。
本实施例序列长度为65的α折叠蛋白质2mqk为实施例,一种基于子种群协同进化的蛋白质结构预测方法,其中包含以下步骤:
1)输入待测蛋白质的序列信息,并从robetta服务器(http://www.robetta.org/)上得到片段库;
2)参数设置:设置种群规模np=100,子种群数量n=5,交叉概率cr=0.5,片段长度l=9,温度因子kt=2,最大迭代次数gmax=1000,并初始化迭代次数g=0;
3)从对各残基位对应的片段库中随机选择片段组装生成初始构象种群p={c1,c2,...,cnp},并将整个种群随机分成n个大小相等的子种群,其中,ci,i={1,2,…,np}为种群p中的第i个构象个体;
4)对种群中的每个构象ci,i∈{1,2,…,np}作如下处理:
4.1)如果构象ci属于第一个子种群,则进行如下操作:
4.1.1)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体ca;
4.1.2)从其余n-2个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cb;
4.1.3)分别从构象个体ca和cb中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.2)如果构象ci属于第二个子种群,则进行如下操作:
4.2.1)根据rosettascore3能量函数计算构象ci所属的子种群的中的每个构象的能量,并选择能量最低的构象记作clbest;
4.2.2)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cc;
4.2.3)分别从构象个体clbest和cc中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.3)如果构象ci属于第三个子种群,则进行如下操作:
4.3.1)根据rosettascore3能量函数计算整个种群中每个构象个体的能量,并按照能量从低到高进行排序;
4.3.2)从排名靠前的np/5种群中随机选择一个构象个体记作cpbest;
4.3.3)从构象ci所属的子种群中随机选择一个与ci和cpbest均不相同的构象个体cd;
4.3.4)分别从构象个体cpbest和cd中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.4)如果构象ci属于其余n-3个子种群,则进行如下操作:
4.4.1)从构象ci所属的子种群中随机选择一个与ci不同的构象ce;
4.4.2)从其余n-1个子种群中随机选择一个子种群,并从该子种群中随机选择一个构象个体cf;
4.4.3)分别从构象个体ce和cf中随机选择两个残基位不同的且长度为l的片段替换构象ci对应位置的片段,生成变异构象个体cmutant;
4.5)随机生成一个0和1之间的小数r,如果r<cr,则从构象ci中随机选取一个长度为l的片段替换变异构象cmutant中对应位置的片段,并进行一次随机片段组装,从而生成测试构象ctrial,否则直接将变异构象进行一次随机片段组装生成测试构象ctrial;
4.6)根据rosettascore3能量函数分别计算测试构象ctrial和ci的能量值,如果ctrial的能量值小于ci的能量值,则ctrial替换ci,否则根据玻尔兹曼概率
5)g=g+1,如果g>gmax,则输出能量最低的构象作为最终预测结构,否则返回步骤4)。
以序列长度为65的α折叠蛋白质2mqk为实施例,运用以上方法得到了该蛋白质的近天然态构象,最小均方根偏差为
以上说明是本发明以蛋白质2mqk为实例所得出的预测结果,并非限定本发明的实施范围,在不偏离本发明基本内容所涉及范围的前提下对其做各种变形和改进,不应排除在本发明的保护范围之外。
- 还没有人留言评论。精彩留言会获得点赞!