一种基于动态抽象凸下界估计的群体蛋白质结构预测方法与流程
本发明涉及一种生物学信息学、智能优化、计算机应用领域,尤其涉及的是一种基于动态抽象凸下界估计的群体蛋白质结构预测方法。
背景技术:
蛋白质结构实验测定方法是结构基因组学研究的主要内容。x射线晶体学是测定蛋白质结构最有效的方法,所能达到的精度是其它方法所不能比拟的,缺点主要是蛋白质晶体难以培养且晶体结构测定的周期较长;多维核磁共振(nmr)方法可以直接测定蛋白质在溶液中的构象,但是由于对样品的需要量大、纯度要求高,目前只能测定小分子蛋白质。总体上,结构实验测定方法主要存在两方面问题:一方面,对于现代药物设计的主要靶标膜蛋白而言,通过实验方法极难获得其结构;另一方面,测定过程费时费钱费力。
在理论探索和应用需求的双重推动下,根据anfinsen法则,利用计算机设计适当的算法,以序列为起点,三维结构为目标的蛋白质结构预测自20世纪末蓬勃发展。从头预测方法实质上就是利用计算机的快速处理能力,利用优化算法在蛋白质构象空间搜索全局最低能量构象解。蛋白质能量模型考虑分子体系成键作用以及范德华力、静电、氢键、疏水等非成键作用,致使其形成的能量曲面极其粗糙,构象对应局部极小解数目随序列长度的增加呈指数增长,属于一类非常难解的np-hard问题。
1994年美国科学家moult发起的两年一届的casp大赛是一个世界性的蛋白质结构预测评比活动,客观地反映了当前蛋白质结构预测领域发展的最新技术水平,被誉为蛋白质结构预测的奥林匹克竞赛。按照2014年casp11最新分类规则,同源建模及折叠识别两种方法统一称为基于模板的蛋白质结构预测方法(templatebasedmodeling,tbm),从头预测改称为自由模板模型预测方法(templatefreemodel,fm)。casp赛事20多年的发展历程表明,对于序列相似度较高的情况(>50%),tbm预测精度能够达到
因此,现有的蛋白质结构预测方法计算代价和预测效率方面存在着缺陷,需要改进。
技术实现要素:
为了克服现有蛋白质结构预测方法的计算代价较高和预测效率较低的不足,本发明提出一种计算代价较小、预测效率较高的基于动态抽象凸下界估计的群体蛋白质结构预测方法。
本发明解决其技术问题所采用的技术方案是:
一种基于动态抽象凸下界估计的群体蛋白质结构预测方法,所述方法包括以下步骤:
1)输入待测蛋白质的序列信息,并从robetta服务器(http://www.robetta.org/)上得到片段库;
2)参数设置:设置种群规模np,交叉概率cr,片段长度l,温度因子kt,斜率控制因子m,初始片段组装次数n,最大迭代次数gmax,并初始化迭代次数g=0;
3)根据输入序列生成二面角φ、ψ、ω分别为-150°、-150°、180°的直链,以此直链为起点启动np条蒙特卡洛轨迹,每条轨迹随机片段组装n次生成一个构象个体,从而生成初始构象种群p={c1,c2,...,cnp},其中,ci,i={1,2,…,np}为种群p中的第i个构象个体;
4)根据rosettasocre3能量函数计算当前种群中每个构象的能量值;
5)根据当前种群中每个构象ci,i∈{1,2,…,np}的碳α原子坐标表示其空间位置坐标
其中,e(ci)为构象ci的能量,
6)对种群中的每个构象ci,i∈{1,2,...,np}执行如下操作:
6.1)将构象ci看作目标构象,并将整个种群随机分成两个大小相等的子种群;
6.2)分别选出这两个子种群中能量最低的构象,分别记作
6.3)从目标构象ci所属的子种群中随机选取一个与ci、
6.4)分别从
6.5)随机生成一个0和1之间的小数r,如果r<cr,则从构象ci中随机选取一个长度为l的片段替换变异构象cmutant中对应位置的片段,并进行一次随机片段组装,从而生成测试构象ctrial;否则直接将变异构象进行一次随机片段组装生成测试构象ctrial;
6.6)计算测试构象ctrial的能量下界估计值
6.7)如果
6.8)如果
6.9)如果ctrial成功替换ci,则根据如下公式计算构象ctrial的抽象凸下界估计支撑向量ltrial,并用ltrial替换ci的支持向量li;
其中,e(ctrial)为构象ctrial的能量,
7)g=g+1,如果g>gmax,则输出能量最低的构象作为最终预测结构,否则返回步骤6)。
本发明的技术构思为:在差分进化算法框架下,基于抽象凸理论,首先,通过计算初始种群中每个构象个体的支撑向量,建立能量函数的下界估计模型;然后,将整个子种群平均划分为两个子种群,从子种群中选择能量最低的构象指导构象变异生成测试构象;其次,计算测试构象的能量下界估计值,根据下界估计信息指导构象的选择;最后,计算被接受的测试构象的支撑向量,并替换对应的目标构象的支撑向量,实现下界估计模型的动态更新。本发明提供一种计算代价较低、预测效率较高的基于动态抽象凸下界估计的群体蛋白质结构预测方法。
本发明的有益效果表现在:通过动态更新能量函数的抽象凸下界估计模型,获取越来越精确的下界估计信息来指导构象的选择过程,避免不必要的能量评价,降低计算代价,进而提高预测效率。
附图说明
图1是基于动态抽象凸下界估计的群体蛋白质结构预测方法对蛋白质1dtja进行结构预测时的构象更新示意图。
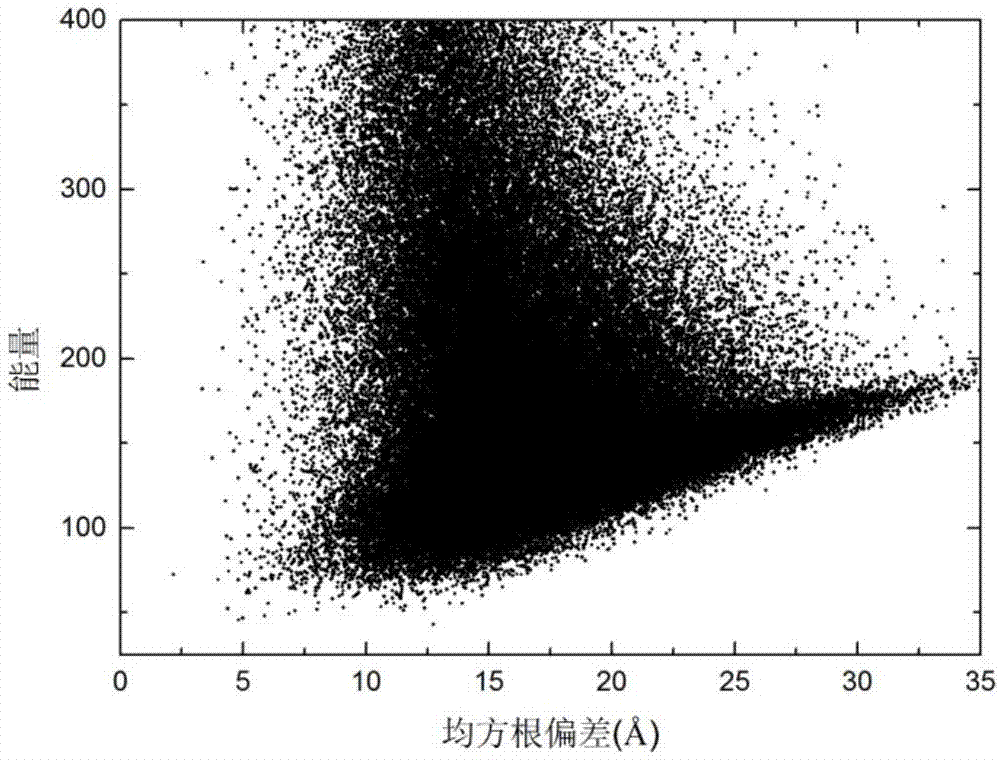
图2是基于动态抽象凸下界估计的群体蛋白质结构预测方法对蛋白质1dtja进行结构预测时得到的构象分布图。
图3是基于动态抽象凸下界估计的群体蛋白质结构预测方法对蛋白质1dtja进行结构预测得到的三维结构图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1~图3,一种基于动态抽象凸下界估计的群体蛋白质结构预测方法,包括以下步骤:
1)输入待测蛋白质的序列信息,并从robetta服务器(http://www.robetta.org/)上得到片段库;
2)参数设置:设置种群规模np,交叉概率cr,片段长度l,温度因子kt,斜率控制因子m,初始片段组装次数n,最大迭代次数gmax,并初始化迭代次数g=0;
3)根据输入序列生成二面角φ、ψ、ω分别为-150°、-150°、180°的直链,以此直链为起点启动np条蒙特卡洛轨迹,每条轨迹随机片段组装n次生成一个构象个体,从而生成初始构象种群p={c1,c2,...,cnp},其中,ci,i={1,2,…,np}为种群p中的第i个构象个体;
4)根据rosettasocre3能量函数计算当前种群中每个构象的能量值;
5)根据当前种群中每个构象ci,i∈{1,2,…,np}的碳α原子坐标表示其空间位置坐标
其中,e(ci)为构象ci的能量,
6)对种群中的每个构象ci,i∈{1,2,...,np}执行如下操作:
6.1)将构象ci看作目标构象,并将整个种群随机分成两个大小相等的子种群;
6.2)分别选出这两个子种群中能量最低的构象,分别记作
6.3)从目标构象ci所属的子种群中随机选取一个与ci、
6.4)分别从
6.5)随机生成一个0和1之间的小数r,如果r<cr,则从构象ci中随机选取一个长度为l的片段替换变异构象cmutant中对应位置的片段,并进行一次随机片段组装,从而生成测试构象ctrial;否则直接将变异构象进行一次随机片段组装生成测试构象ctrial;
6.6)计算测试构象ctrial的能量下界估计值
6.7)如果
6.8)如果
6.9)如果ctrial成功替换ci,则根据如下公式计算构象ctrial的抽象凸下界估计支撑向量ltrial,并用ltrial替换ci的支持向量li;
其中,e(ctrial)为构象ctrial的能量,xttrial,t∈{1,2,...,3l}为构象ctrial空间位置坐标的第t维元素,
7)g=g+1,如果g>gmax,则输出能量最低的构象作为最终预测结构,否则返回步骤6)。
本实施例序列长度为76的α/β折叠蛋白质1dtja为实施例,一种基于动态抽象凸下界估计的群体蛋白质结构预测方法,其中包含以下步骤:
1)输入待测蛋白质的序列信息,并从robetta服务器(http://www.robetta.org/)上得到片段库;
2)参数设置:设置种群规模np=50,交叉概率cr=0.5,片段长度l=9,温度因子kt=2,斜率控制因子m=10000,初始片段组装次数n=1000,最大迭代次数gmax=1000,并初始化迭代次数g=0;
3)根据输入序列生成二面角φ、ψ、ω分别为-150°、-150°、180°的直链,以此直链为起点启动np条蒙特卡洛轨迹,每条轨迹随机片段组装n次生成一个构象个体,从而生成初始构象种群p={c1,c2,...,cnp},其中,ci,i={1,2,…,np}为种群p中的第i个构象个体;
4)根据rosettasocre3能量函数计算当前种群中每个构象的能量值;
5)根据当前种群中每个构象ci,i∈{1,2,…,np}的碳α原子坐标表示其空间位置
坐标
其中,e(ci)为构象ci的能量,
6)对种群中的每个构象ci,i∈{1,2,...,np}执行如下操作:
6.1)将构象ci看作目标构象,并将整个种群随机分成两个大小相等的子种群;
6.2)分别选出这两个子种群中能量最低的构象,分别记作
6.3)从目标构象ci所属的子种群中随机选取一个与ci、
6.4)分别从
6.5)随机生成一个0和1之间的小数r,如果r<cr,则从构象ci中随机选取一个长度为l的片段替换变异构象cmutant中对应位置的片段,并进行一次随机片段组装,从而生成测试构象ctrial;否则直接将变异构象进行一次随机片段组装生成测试构象ctrial;
6.6)计算测试构象ctrial的能量下界估计值
6.7)如果
6.8)如果
6.9)如果ctrial成功替换ci,则根据如下公式计算构象ctrial的抽象凸下界估计支撑向量ltrial,并用ltrial替换ci的支持向量li;
其中,e(ctrial)为构象ctrial的能量,
7)g=g+1,如果g>gmax,则输出能量最低的构象作为最终预测结构,否则返
回步骤6)。
以序列长度为76的α/β折叠蛋白质1dtja为实施例,运用以上方法得到了该蛋白质的近天然态构象,均方根偏差为
以上说明是本发明以蛋白质1dtja为实例所得到的结果,并非限定本发明的实施范围,在不偏离本发明基本内容所涉及范围的前提下对其做各种变形和改进,不应排除在本发明的保护范围之外。
- 还没有人留言评论。精彩留言会获得点赞!