一种促进胰岛素等大分子药物的口服吸收的吸收促进剂的制作方法
本发明属于医药生物领域,具体而言,涉及一种促进胰岛素等大分子药物的口服吸收的吸收促进剂。
背景技术
治疗性生物大分子如蛋白质和多肽已成为有效的治疗型制剂(1),但是由于其口服时跨粘膜运输的效率较低,影响了药物的输送,所以这些生物大分子在慢性疾病中的使用受到限制(2),因此如何跨越粘膜屏障安全有效地递送蛋白质和多肽等治疗剂仍然是这一领域的主要挑战。而肠道中的肠腔酶降解使这一过程变得更加复杂(3),目前,虽然同时提供对蛋白质和多肽的结构保护剂以及粘膜吸收促进剂的许多策略已经取得了一些进展(4),但是这些技术中也都存在实际的问题。
两亲性聚合物在超过临界胶束浓度(cmc)的浓度下通过自我聚集固有地形成胶束,并且在我们的前期工作中,这些胶束已成功用作非口服给药时的纳米药物载体(6,7)。虽然它们在口服给药中的性能还没有被广泛研究,但是聚合物胶束能够非常有效地装载蛋白质和肽类药物(cn104288758b),保护它们免于酶降解的特性。
蛋白质和多肽是两亲性分子,受其浓度和溶剂条件的影响,会形成各种形式的聚集体(8),假设两亲聚合物分子可能通过静电和疏水相互作用以及氢键与蛋白质和多肽具有很强的亲和力(9),因此,我们在本研究中选择了制药工业中常用的不同化学成分的两亲性聚合物,在这些两亲性聚合物中,我们最感兴趣的是聚乙二醇-磷脂酰乙醇胺(peg-pe)两亲性分子形成的胶束。peg-pe是一种可生物降解的两亲性聚合物,柔性聚乙二醇链(peg)为亲水性壳,磷脂酰乙醇胺(pe)结构作为细胞膜的组成部分,起到疏水性核心作用。
我们前期的研究表明,某些水溶性药物可以成功地通过在水溶液中以一个简单的一步自组装的方式加载到自组装形成peg-pe胶束中(9,10),这些载药胶束通过拆卸和插入细胞膜将它们的有效载荷的药物卸载到细胞膜上,从而导致膜流动性的增加并加速药物的细胞内转运(11)。最近,我们发现peg-pe胶束具有天然分子伴侣的特性,可以帮助变性胰岛素重新折叠并防止胰岛素聚集(12)。在这种情况下,peg-pe胶束提供亲水性纳米笼,能够容纳胰岛素a和b链,并促进两条链正确折叠。根据形成的纳米笼的空间(12),peg-pe胶束可以容纳分子量小于20kd的蛋白质分子。
基于上述对peg-pe胶束的结构和功能的研究,有必要探索peg-pe两亲性聚合物作为口服递送蛋白质和多肽治疗剂的吸收促进剂的进一步研究。
技术实现要素:
本发明首先涉及一种peg-dspe分子在制备促进大分子药物口服吸收的吸收促进剂中的应用,所述的peg-dspe分子的亲水嵌段为链分子量1000~5000的聚乙二醇(peg),疏水核心区为1,2-二硬脂酰-sn-甘油基-3-磷酸乙醇胺(dspe),所述的吸收促进剂的功能是:
(1)促进大分子药物快速通过消化道粘液层;
和/或(2)促进大分子药物快速通过消化道上皮细胞间渗透通路进行吸收。
优选的,所述的peg-dspe分子的亲水嵌段为链分子量为2000的聚乙二醇(peg)。
所述的大分子药物的分子量为0.5kda~70kda。
优选的,所述的大分子药物的分子量为0.5~50kda;
更优选的,所述的大分子药物的分子量为0.5~20kda;
最优选的,所述的大分子药物的分子量为0.5~10kda。
所述的大分子药物为蛋白、肽、糖或聚糖、抗生素或化学药物;
优选的,所述的大分子药物为胰岛素、glp-1、鲑降钙素、奥曲肽、环孢菌素、雷帕霉素、细胞因子(干扰素等)。
本发明还涉及一种口服给药后生物利用度大于3%的大分子口服制剂,其包含
(1)作为吸收促进剂的peg-dspe,所述的peg-dspe分子的亲水嵌段为链分子量1000~5000的聚乙二醇(peg),疏水核心区为1,2-二硬脂酰-sn-甘油基-3-磷酸乙醇胺(dspe);
(2)治疗有效量的大分子药物;
所述的peg-dspe与大分子药物的摩尔比为2~10:1。
优选的,所述的peg-dspe分子的亲水嵌段为链分子量为2000的聚乙二醇(peg);
所述的药用大分子药物为分子量0.5kda~70kda的药用大分子药物;
优选的,所述的大分子药物的分子量为0.5~50kda;
更优选的,所述的大分子药物的分子量为0.5~20kda;
最优选的,所述的大分子药物的分子量为0.5~10kda。
优选的,所述的大分子药物为蛋白、肽、糖或聚糖、抗生素或化学药物;
更优选的,所述的大分子药物为胰岛素、glp-1、鲑降钙素、奥曲肽、环孢菌素、雷帕霉素、细胞因子、干扰素等。
本发明还涉及peg-dspe分子在制备口服给药后生物利用度大于3%的大分子药物口服制剂中的应用,
所述的peg-dspe分子的亲水嵌段peg的链分子量为1000~5000,优选的,为peg2000-dspe;
所述的大分子药物为分子量0.5kda~70kda的蛋白、肽、糖或聚糖、抗生素或化学药物,优选的,所述的大分子药物为胰岛素、glp-1、鲑降钙素、奥曲肽、环孢菌素、雷帕霉素、细胞因子、干扰素等。
本发明还涉及一种口服给药的药物制剂,所述的药物制剂包括:
(1)治疗有效量的胰岛素、glp-1或鲑降钙素;
(2)作为吸收促进剂的peg2000-dspe;
(3)必要的其他药用辅料。
所述的peg2000-dspe与胰岛素、glp-1和鲑降钙素的摩尔比分别为:5~10:1、3~10:1和2~10:1;
优选的,所述的peg2000-dspe与胰岛素、glp-1和鲑降钙素的摩尔比分别为:5:1、3:1和2:1。
本发明还涉及peg2000-dspe分子在制备调节zo-1蛋白的细胞膜-胞浆分布比例的制剂中的应用,所述的调节为,诱导zo-1分子从细胞膜上移动至胞浆中;且所述的诱导效应为可逆的,即添加peg2000-dspe分子时诱导zo-1分子从细胞膜上移动至胞浆中,洗去peg2000-dspe分子时zo-1回复至膜上。
本发明还涉及peg2000-dspe分子在制备阻断紧密连接蛋白-2(claudin-2)的胞外环状结构域的相互作用的制剂中的应用,所述的claudin-2胞外环状结构域为氨基酸序列为seqidno.1所示的的eld2结构域,所述的阻断为可逆的,即添加peg2000-dspe分子时阻断,洗去peg2000-dspe分子时恢复。
seqidno.1:wnlhgilrdfysplvpdsmkfeigek。
本发明还涉及peg2000-dspe分子在制备调节细胞间紧密连接的制剂中的应用,所述的调节为,
(1)通过降低zo-1骨架分子的细胞膜-胞浆分布比例,从而减少细胞间的紧密连接数量,
(2)阻断紧密连接蛋白-2(claudin-2)的胞外环状结构域的相互作用,从而阻断细胞间紧密连接的形成;
且所述的调节为可逆的,即添加peg2000-dspe分子产生调节效果,洗去peg200-dspe分子恢复。
附图说明
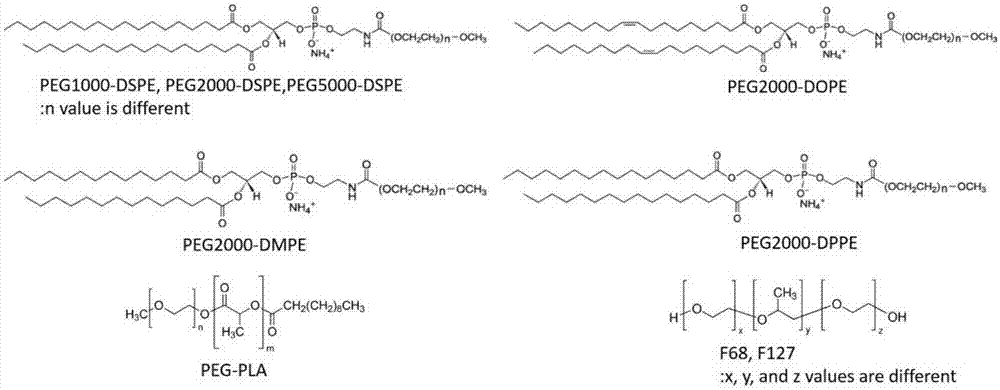
图1、两亲性聚合物的分子结构。
图2、peg2000-dspe是一种安全有效的渗透增强剂;不同链长的pegs-pes(a)或其他类型的两亲性聚合物(b)在不同浓度下处理caco-2单细胞层8小时后的细胞毒性(n=5);两亲性聚合物对caco-2单细胞层的teer值的影响(c,d);fd10(e,f)或fd70(g)与两亲性聚合物共同孵育caco-2单细胞层后,透过单细胞层的累计量的时间函数,阳性对照为常用的化学渗透促进剂egta和sds。数据表示来自四次重复实验(n=4)的平均值±sd。
图3、peg2000-dspe处理单层细胞增加右旋糖苷和葡聚糖分子的细胞旁渗透通量(3kda为蓝色荧光标记的右旋糖酐,10kda为alexafluor647标记的葡聚糖)(a:caco-2细胞单层,b:mdck细胞单层)。洗涤单层细胞并转移至hanks平衡盐溶液(hbss)中,轻轻吸出上室平衡液,并用200μl含有23.5μg蓝色荧光标记的3kda葡聚糖,78.1μgalexafluor647标记的10kda葡聚糖的hbss缓冲液换液,在37℃下连续培养单细胞层,120分钟后从基底室取出100μl样品,使用荧光板读数器(synergyht;bio-tekinstruments,winooski,vt)测定这些样品的荧光。
图4、peg2000-dspe在生理范围内调节药物通过细胞旁路转运图,在peg2000-dspe处理(a)或不处理(b)的情况下,不同分子量的荧光标记葡聚糖(fds)的caco-2单细胞层细胞旁路的累积转运曲线;peg2000-dspe处理或不处理与fds的表观渗透系数(papp)之间的关系(c);在sds(d)和egta(e)存在下,fds在caco-2单细胞层上的累积转运情况。在caco-2单细胞层中peg2000-dspe处理增加fd10细胞旁路转运的持续时间:前四小时fd10单独处理caco-2单细胞层,然后加入peg2000-dspe共同处理四小时(f);或前四个小时fd10与peg2000-dspe共同处理caco-2单细胞层,然后洗去peg2000-dspe后,fd10单独与caco-2单细胞曾孵育四小时(g),两种处理结果显示fd10透过caco-2单细胞层的转运速率的差异。fds:具有不同平均分子量的异硫氰酸荧光素标记的葡聚糖(fd4,fd10,fd20,fd40和fd70)。
图5、peg2000-dspe选择性调节zo-1动态分布。通过透射电子显微镜观察紧密连接的超微结构变化:使用1mmpeg2000-dspe处理2小时的caco-2细胞(a)和大鼠空肠上皮(b);1mmpeg2000-dspe处理2小时后,caco-2细胞单层的zo-1,e-cad和actin(肌动蛋白)的分布的变化(c);用1mmpeg2000-dspe处理2小时caco-2细胞后,将细胞分离成细胞质成分和总成分,并对zo-1、actin和e-cad蛋白进行免疫印迹定量以检测zo-1,actin和e-cad的变化结果(d);(e)表达mcherryzo-1的caco-2细胞连续成像4小时图(成像期间以10分钟间隔对代表性区域拍照,未处理的对照细胞在上排,peg2000-dspe处理的细胞在下排);(f)未处理的对照细胞(上图)和peg2000-dspe处理的细胞(下图)的mcherryzo-1细胞膜结构变化图。
图6、peg2000-dspe与棕榈酰化的claudin-2细胞外环结构域(pal-eld2:n末端和c末端均被棕榈酸修饰)相互作用。(a)将所有样品稀释,点在盖玻片上,并通过共聚焦显微镜观察:分别用dio或dii标记的脂质体(左图)或pal-eld2插入的脂质体(中图),以及peg2000-dspe处理后pal-eld2插入的脂质体(右图)。(b)通过在405nm波长处的光吸收测量的加入不同浓度的peg2000-dspe以阻断pal-eld2插入的脂质体的聚集效应,数字表示pal-eld2与peg2000-dspe的摩尔比。(c)将pal-eld2与不同浓度的peg2000-dspe一起温育,通过cd色谱法测量peg2000-dspe对pal-eld2的二级结构的影响。(d)通过与peg2000-dspe,peg2000-dsg和peg2000-l-dsg一起孵育的caco-2细胞单层的fd10透过的累积量作为时间的函数。
图7、peg2000-dsg及peg2000-l-dsg的化学结构。
图8、装载蛋白质与多肽药物的peg2000-dspe胶束的理化性质表征:(a)装载有胰岛素的peg2000-dspe胶束(m-ins);(b)装载sct的peg2000-dspe胶束(m-sct)和(c)装载有glp-1的peg2000-dspe胶束(m-glp-1)的透射电子显微镜(tem)图像(采用1%乙酸双氧铀染色并通过电子显微镜观察);peg2000-dspe与胰岛素(d)、sct(e)和glp-1(f)的摩尔比对包封效率的影响;peg2000-dspe胶束在313k水中的微量热滴定:将总共3.3mm的peg2000-dspe胶束逐渐加入到含有每种药物溶液的反应池中(箭头表示滴定方向):胰岛素(g),sct(h)和glp-1(i)。
图9、peg2000-dspe胶束装载蛋白质与多肽药物后的装载改变了药物的聚集结构:pbs缓冲液(a)中的胰岛素和peg2000-dspe胶束中的胰岛素(b)的沉降系数分布图;不同ph条件下或peg2000-dspe胶束(m-ins)中的胰岛素的二级结构(c,远紫外光谱)和三级结构(d,近紫外光谱)图谱;(e)peg2000-dspe胶束装载的sct(m-sct)溶液或游离sct溶液的代表性圆二色谱图;(f)peg2000-dspe胶束装载的glp-1(m-glp-1)溶液或glp-1溶液的代表性圆二色谱图。
图10、peg2000-dspe胶束装载的胰岛素增强了其在小肠粘液扩散和小肠上皮渗透能力。(a)通过frap(光漂白后荧光恢复)技术,在离体分离的大鼠小肠粘液中研究f-ins、m-ins或l-ins的自由扩散;(b)从图a中光漂白后荧光恢复曲线计算f-ins、m-ins和l-ins中快速移动药物分子的比例(themobilefraction)及荧光值恢复到初始值一半的时间(t1/2);(c)通过caco-2细胞旁路转运的胰岛素累积转运量;(d)共聚焦激光扫描显微镜(clsm)图像显示,m-ins(tritc-胰岛素(红色)或fitc-peg2000-dspe(绿色))孵育的caco-2单细胞层的药物吸收,图片显示为纵轴依次增加5μm的四个光学切片,单独tritc-胰岛素溶液(红色)作为对照;在十二指肠给予f-ins(e,胰岛素为fitc标记)或m-ins(f,胰岛素为fitc标记)后在正常大鼠的小肠的不同节段和相应肠系膜中吸收的胰岛素的clsm图像;(g)在大鼠模型中十二指肠给予荧光标记的m-ins(fitc标记胰岛素和alexa660标记peg2000-dspe)2小时后获得的大鼠空肠的clsm图像。
图11、peg2000-dspe聚合物的细胞旁转运,(a)与m-ins共孵育30min后的caco-2细胞的共焦显微镜图像(tritc-胰岛素,红色;fitc-peg2000-dspe,绿色);(b)caco-2单层细胞与m-ins共孵育30分钟后,洗去m-ins,结果显示在细胞膜上仅能观察到fitc-peg2000-dspe荧光。
图12、大鼠十二指肠给予m-ins2h后,小肠不同部位(十二指肠,空肠和回肠)吸收胰岛素的情况(免疫组化结果)。
图13、糖尿病大鼠十二指肠(dd)给予m-ins或f-ins,或通过皮下注射(sc)给予f-ins后的大鼠血糖水平(a)和血浆胰岛素浓度(b),正常大鼠十二指肠给予m-sct或f-sct,或皮下注射(sc)给予f-sct后的大鼠血钙水平(c)和血浆中鲑降钙素浓度(d)(平均值±sd,n=4)。
具体实施方式
材料和方法
一、材料
1,2-二硬脂酰-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)-2000](铵盐)(peg2000-dspe)、1,2-二肉豆蔻酰基-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)-2000](铵盐)(peg2000-dmpe)、1,2-二油酰基-sn-甘油基-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)-2000](铵盐)(peg2000-dope)、1,2-二棕榈酰-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)-2000](铵盐)(peg2000-dppe)、1,2-二硬脂酰-sn-glycero-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)-1000](铵盐)(peg1000-dspe)、1,2-二硬脂酰-sn-甘油-3-磷酸乙醇胺-n-[甲氧基(聚乙二醇)-5000](铵盐)(peg5000-dspe)都购自avantipolarlipids(alabaster,al)。
sds、聚(乙二醇)甲基醚-嵌段-聚(d,l-丙交酯)-嵌段-癸烷(peg-pla)、
鲑鱼降钙素(sct)和胰高血糖素样肽-1(glp-1)获自上海延长生化开发有限公司(中国上海)
sct的氨基酸序列(seqidno.2):cysserasnleuserthrcysvalleuglylysleuserglngluleuhislysleuglnthrtyrproargthrasnthrglyserglythrpro-nh2(二硫键位置:cys1-cys7)
seqidno.2:csnlstcvlgklsqelhklqtyprtntgsgtp
glp-1的氨基酸序列(seqidno.3):
hisaspgluphegluarghisalagluglythrphethrseraspvalsersertyrleugluglyglnalaalalysglupheilealatrpleuvallysglyarggly
seqidno.3:hdeferhaegtftsdvssylegqaakefiawlvkgrg
猪胰岛素(30iu/mg)购自wanbangbiochemicalco.,ltd。(江苏,中国)。
pal-eld2(seqidno.1:wnlhgilrdfysplvpdsmkfeigek,n末端和具有棕榈酸修饰的c末端)由lifetein(中国北京)合成。
其他试剂都是分析纯等级。
二、细胞株及动物
caco-2细胞获自中科院生物化学与细胞生物学研究所(中国上海)。
表达mcherryzo-1和gfp-actin的caco-2细胞系和mdck细胞由jerroldr.turner博士(brighamandwomen’s医院,美国)提供。
动物实验经中国科学院生物物理研究所机构动物护理和使用委员会批准进行。
重量为220±20g的雄性sprague-dawley大鼠由vitalriverlaboratoryanimaltechnologyco.ltd。(中国北京)提供。
三、细胞培养
caco-2细胞,人结肠腺癌细胞,传代数36-42,在37℃,5%co2的培养箱中培养。使用含有10%胎牛血清,10,000u/ml青霉素和10,000μg/ml链霉素的modifiedeagle's培养基(mem)将细胞维持在t-75培养瓶中。生长培养基每隔一天更换一次,细胞汇合至80至90%时,使用0.25%胰蛋白酶/乙二胺四乙酸溶液进行传代。
四、细胞活性实验
使用mtt测定评估聚合物材料对caco-2细胞的细胞毒性:将caco-2细胞以1.0×104个细胞/孔的密度接种到96孔板上并培养7天。将细胞与一系列浓度的聚合物材料在hbss中于37℃温育8小时,然后进行mtt测定。测试一式三份进行,数据通过与对照比较表示为活细胞的百分比。
五、跨上皮电阻(teer)和标志物细胞旁通量的测量
teer测量:caco-2细胞单层在含预处理过的聚碳酸酯滤孔costartranswell12孔/板(corningcostarcorp.)上培养,并在接种后约21天后使用(teer值在600-800ω·cm2范围内)。然后,将聚合物材料分别置于transwell的供体室中,并使用millicell电阻系统(milliporecorp.,bedford,ma,usa)测量与细胞单层紧密度相关的teer变化。
标志物细胞旁通量的测量:在caco-2单层中进行细胞旁标志物通量分析,将具有或不具有聚合物材料的1mg/mlfitc标记的葡聚糖(分别具有4,10,40和70kda的fd4,fd10,fd40,fd70)添加至顶侧,并且在预定时间收集基底外侧样品,使用荧光板读数器测定这些样品的荧光,从每日制备的标准曲线计算摩尔通量。为了检查peg2000-dspe诱导的渗透增强的持续时间,将peg2000-dspe去除后的fd10渗透性与peg2000-dspe诱导的fd10渗透性进行比较。
六、胰岛素转运和共聚焦激光扫描显微镜(clsm)可视化研究
caco-2细胞单层在含预处理过的聚碳酸酯滤孔costartranswell12孔/板(corningcostarcorp.)上培养,并在接种后约21天后用于转运实验。使用游离形式的胰岛素溶液作为对照,在caco-2细胞单层中研究了测试的胶束(1mg/ml,2ml)对增强胰岛素的细胞旁转运的影响。以预定的时间间隔(0分钟,20分钟,40分钟,60分钟,80分钟,100分钟,120分钟)从接收室收集样品(100μl),并使用hplc测量胰岛素浓度。
根据以下等式计算胰岛素或fd的表观渗透性(papp,cm/s):
papp=[dqt/dt]×[1/(a×c0)]
其中dqt/dt是胰岛素累积量或fd转运量(ng)与时间(s)之间线性关系的斜率,a是细胞单层面积(cm2),c0是供体室胰岛素或fd的初始浓度(ng/cm3)。
为了使胰岛素和peg2000-dspe聚合物转运可视化,用双荧光标记的m-ins(tritc标记的胰岛素和fitc标记的peg2000-dspe)处理2小时后的单细胞层用温pbs洗涤两次并用4%(w/v)的多聚甲醛溶液固定。然后在共聚焦激光显微镜下观察固定的细胞(clsm,olympus,japan)
七、免疫荧光染色
将单细胞层用含1%多聚甲醛的ph7.4的pbs在室温下固定30分钟,pbs中洗涤三次并在含有50mmnh4cl的pbs中孵育15分钟后,将单细胞层用含有1%bsa和0.05%皂苷的pbs渗透,然后与添加一抗的含有1%bsa和0.05%皂苷的pbs溶液(zo-1,肌动蛋白,e-钙粘蛋白,occludin,claudin-1,claudin-2,claudin-3,claudin-4,claudin-5,claudin-7,claudin-8,claudin-10)(invitrogen,usa)在4℃下孵育过夜;用含有1%bsa和0.05%皂苷的pbs溶液洗涤三次;并与适当的alexafluor488-或594-缀合的二抗(invitrogen,usa)一起温育,孵育后再洗涤三次,将单层细胞在水中漂洗并固定在prolongdiamondantifademountant(invitrogen,usa)中。
八、蛋白质提取和western免疫印迹实验
总蛋白质提取,在peg2000-dspe处理后,用冰冷的pbs轻轻洗涤caco-2细胞两次,然后用裂解缓冲液覆盖细胞。在冰上进行裂解反应30分钟,每5分钟进行晃动一次。随后,将样品以10,000rpm离心5分钟,收集上清液用于随后的蛋白质印迹分析。为了检测peg2000-dspe处理后亚细胞水平下的蛋白质表达,通过fractionprep细胞分级试剂盒(biovison,mountainview,ca,usa)提取细胞溶质蛋白质级分。首先,用冰冷的pbs收集细胞,并以700g离心5分钟。除去上清液后,将细胞沉淀重悬于细胞溶质提取缓冲液-混合物中,并在冰上温育20分钟,每5分钟轻轻敲一下,将样品以700g离心10分钟,收集上清液作为胞质部分。
蛋白质免疫印迹:如上所述制备样品,在95℃煮沸5分钟后,将等分试样在sds-paga凝胶上分离,并转移到聚偏二氟乙烯膜上。用针对gapdh,zo-1,密蛋白-2和occludin的一抗(invitrogen)探测目标靶蛋白。将膜在5%脱脂奶粉中在tris缓冲盐水(tbs)中封闭,在含有0.05%tween20(tbs-t)的tbs中洗涤,并与含有1%脱脂奶粉的tbs中的一抗在4℃温育过夜。用tbs-t洗涤膜,与合适的辣根过氧化物酶缀合的二抗一起孵育,并在检测之前用tbs-t洗涤。在tanon-5200化学发光成像系统(tanonscienceandtechnology)上检测发光信号。使用imagej软件(nationalinstitutesofhealth,bethesda,md)进行密度测定。
九、活细胞成像
表达caco-2的mcherryzo-1单层在补充有20mmhepes,ph7.4的fluorobritedmem培养基中成像。使用由metamorph软件控制的leicadmi6000显微镜进行成像。全部z-堆叠每10分钟成像,持续成像4小时;获取图像并使用变形法进行最大强度投影。
十、脂质体的制备
通过挤出方法获得大的单层囊泡(luv)。将egg-pc(卵磷脂)和胆固醇以设计的摩尔比溶解在氯仿中,然后在氮气下蒸发溶剂并进一步在真空下干燥过夜。加入hepes缓冲液(10mm)以使脂质膜水化,然后在55℃下加热30分钟。剧烈摇动混合物以获得多层脂质体(mlv)。为了获得100nmluv,通过挤出(avantipolar)使mlv悬浮液通过400nm聚碳酸酯膜过滤器10遍,然后过100nm过滤器(whatman)缩小脂质体粒径。通过在氯仿中溶解不同的脂质(包括dio)制备荧光脂质体(dio-或dii-标记的),棕榈酰化的eld2肽标记的脂质体(pal-eld2脂质体)和荧光标记的pal-eld2脂质体(dio-或dii-标记的pal-eld2脂质体)。
十一、载药peg2000-dspe胶束的制备
溶解蛋白质药物(sct或glp-1),并将peg2000-dspe分别悬浮在双蒸水中。所得药物和peg2000-dspe储备溶液浓度都为5mm。然后,按照目标peg2000-dspe:药物比率将peg2000-dspe和药物储备溶液在圆底混合物中混合,将混合物在60℃温育30分钟。孵育后,使用0.22μm无菌注射器膜过滤除菌所得溶液。将过滤的溶液储存在4℃直至使用。使用相同的步骤,不添加药物制备空胶束。为了制备负载胰岛素的胶束,将胰岛素粉末悬浮在双蒸水中并与peg2000-dspe储备溶液混合至所需摩尔比。将混合物在60℃温育30分钟并轻轻摇动。温育后,将所得悬浮液离心。peg2000-dspe胶束中的药物通过rp-hplc方法使用c18柱测定。此外,为了在以下测试中观察和评估胰岛素和peg2000-dspe,按照以下文献中描述的方法合成fitc标记的peg2000-dspe,tritc标记的peg2000-dspe,fitc标记的胰岛素和tritc标记的胰岛素。文献(11,31)。通过用1.5kda离心过滤器离心除去未缀合的染料。
十二、药物包封率的测量
通过超滤确定药物在胶束中的包封效率。将500μl胶束制剂(包括装载在peg2000-dspe胶束中的sct和glp-1)加入amicon超离心过滤装置(mwco50,000,millipore)中,然后在4℃下以12000g离心10分钟。过滤中的游离药物浓度由bicinchoninicacidproteinassaykit定量。为了确定胰岛素包封效率,在离心管中加入500μl胶束悬浮液,然后在25℃下以12000g离心10分钟。通过高效液相色谱(hplc)在上清液中测定胶束中的胰岛素量,并通过与加入的初始胰岛素的比率计算包封效率。
十三、等温滴定方法
使用itc在323k(microcal,llc,northampton,ma,usa)测量peg2000-dspe结合药物的热流。在每次滴定中,将200μl反应池加载不同的药物,并使用装载所需浓度的药物和peg2000-dspe的胶束40μl的自动注射器,使用400rpm的搅拌速度,进行38次连续1μl等分试样的序列。使用peg2000-dspe滴定到纯水中进行对照实验以校正稀释和混合的热效应。使用microcaloriginv.7.0软件(originlabcorp.,northampton,ma,usa)分析量热数据并将其转换成焓变化。实验数据分为单点结合模型,结合常数(kb),结合数(n),焓(△h),熵(△s)和结合自由能(△g)用软件计算。
十四、cd光谱检测
通过圆二色谱(cd)测定peg2000-dspe胶束或游离药物溶液中的肽二级结构。使用chirascanspectropolarimeter(appliedphotophysics,uk)在远紫外(190nm-260nm)或近紫外(260nm-300nm)区域使用0.1cm光程长度在1nm的单元中记录光谱。灯罩用氮气吹扫,平均获得三次扫描。同时,还获得了适当的对照溶液cd光谱并从相应的蛋白质光谱中减去。
十五、粘液扩散研究
用光漂白后的荧光恢复(frap)测量载有胰岛素的peg2000-dspe胶束和装载胰岛素的脂质体在大鼠粘液中的扩散。所有测量均在室温(25℃)下进行。实验在玻璃底室中进行,将稀释的溶液(10μl)加入到100μl新鲜大鼠粘液中并在测量前温育15分钟。使用共聚焦激光扫描显微镜进行frap。488nm用于样品漂白和荧光激发。漂白和恢复成像的光源设置分别是最大激光功率的100%和5%。从整个区域中选择一个圆圈进行漂白,记录漂白区域的时间序列图像。记录漂白区域的荧光强度。通过以下公式预测穿透的fitc-胰岛素的量:mf=(imax-i0)/(1-i0),其中imax是光漂白后的恢复值,i0是光漂白后的值。
十六、跨肠上皮运输
通过clsm研究了m-ins(胰岛素载体peg2000-dspe胶束)在小鼠不同节段的小肠中的胰岛素吸收增强作用。合成fitc标记的胰岛素和tritc标记的peg2000-dspe并用于制备荧光m-ins。十二指肠给药后2小时后处死大鼠,并且基于它们相对于胃和结肠的解剖学位置解剖并鉴定的相应肠段(十二指肠,空肠,回肠和肠系膜)。接下来,用磷酸盐缓冲盐水(pbs)洗涤切开的肠段,在多聚甲醛中固定,并进行冷冻切片处理。通过在载玻片上伸展来制备对应于小肠段的肠系膜样本。并在clsm下检查标本。
十七、紧密连接的超微结构观察
使用透射电子显微镜(tem)检测peg2000-dspe对caco-2细胞单层的超微结构水平的影响。用peg2000-dspe处理caco-2细胞单层2小时,用pbs洗涤,然后固定在3.7%多聚甲醛中。随后,将细胞单层样品固定在四氧化锇中并使用梯度醇浴(25%,50%,75%和100%)脱水。然后将脱水的样品包埋在正丁基树脂中过夜以在70℃下聚合,用金刚石刀切割超薄切片,然后加载到tem网格上并通过tem检查。鉴定并记录相邻caco-2细胞之间的连接复合物。
还通过tem研究了peg2000-dspe的体内tj调节。在该研究中,将peg2000-dspe给予过夜禁食的大鼠。2小时后将动物处死,解剖空肠节段,用等渗盐水冲洗,并固定在3.7%多聚甲醛中。使用前述相同的方法将固定的组织切成较小的片并进行tem研究。
十七、测定降血糖功效
通过连续3天每天以65mg/kg体重的剂量在腹膜内注射链脲佐菌素(stz,溶于ph4.5的10mm柠檬酸盐缓冲液中)诱导雄性sprague-dawley大鼠(180-200g)糖尿病模型。在stz处理后两周,当空腹血糖水平高于300mg/dl时,成功诱导大鼠糖尿病。将动物随机分成4组,每组4只。将糖尿病大鼠禁食但不禁水过夜。然后通过十二指肠内给药给动物施用不同的样品,包括游离胰岛素溶液(100iu/kg),pbs溶液和m-ins(100iu/kg)。通过皮下(sc)注射给一组大鼠施用胰岛素溶液(5iu/kg)。在给药之前和给药后的不同时间从大鼠的眼眶静脉丛收集血液样品,并用血糖仪测试。为了分析血清胰岛素水平,将血液样品离心(3000rpm,5分钟),随后使用胰岛素elisa试剂盒(r&dsystem,inc.,mn,usa)定量。使用下式计算十二指肠内给药后m-ins的相对生物利用度(fr%)。
实施例1、两亲性聚合物作为渗透促进剂
1、生物安全性
因为吸收差和生物利用度低,蛋白质和多肽治疗剂通过口服给药时,通常需要吸收促进剂来促进药物的吸收(13)。最常用的吸收促进剂是瞬时破坏上皮细胞膜的小分子化合物。虽然这确实会促进药物口服递送,但这些吸收促进剂可能会在局部或全身导致严重的毒性反应(14)。与此相对的,两亲性聚合物(图1)可能同样具有吸收促进剂的功能,并且由于两亲性聚合物的生物安全性相对较高,我们推测两亲性聚合物可以在没有细胞毒性的基础上增强药物的跨小肠上皮细胞递送。
为了检验这一假设,我们首先检测了两亲性聚合物(图2)对小肠上皮细胞的细胞毒性。我们选择的两亲性聚合物的亲水嵌段为不同链长的聚乙二醇(peg),疏水核心区为1,2-二硬脂酰-sn-甘油基-3-磷酸乙醇胺(dspe)、1,2-二油酰基-sn-甘油基-3-磷酸乙醇胺(dope)或1,2-二肉豆蔻酰-sn-甘油-3-磷酸乙醇胺(dmpe);在0.1mm至10mm浓度下测试了上述两亲性聚合物(图2a)以及其它常用吸收促进剂(图2b)对细胞活力的影响。
结果显示,与其他测试材料不同,peg-dspe对caco-2细胞活力或生长没有显著影响。我们还测试了两亲性聚合物对跨上皮电阻(teer,一种上皮屏障功能的敏感度量)的影响。除了peg2000-dspe和peg5000-dspe的以外,所有测试的材料显著地降低了caco-2细胞层的屏障功能(图2c-d)。
通过细胞旁路途径运输离子和大分子的流道可以分为通过“孔道(pore)”途径和通过“泄漏(leak)”途径两种方式完成(引用turner2009,naturereviewimmunologyandshen,weber,andturnerannualreviewsofphysiology)。对teer没有影响表明化合物没有改变通过“孔道”途径的流道通量,但对teer没有影响不能排除对“渗漏”途径也没有影响。为了评估“渗漏”途径通量,我们测试了不同类型的吸收促进剂对10kda葡聚糖(fd10)转运的影响。如预期的那样,破坏细胞连接的egta、降低细胞活力的化合物sds、以及peg2000-dmpe等都显著增强fd10的细胞旁通透性(通过泄漏途径的跨细胞输送)(p<0.05),相反,peg-dspe型聚合物(peg2000-dspe和peg5000-dspe)相对较小地增加了fd10通量(图2e,f)。同时,为了评估这些吸收促进剂对目标药物分子大小的选择性,进一步测量70kda葡聚糖(fd70)的细胞旁通透性(正常情况下,70kda葡聚糖因分子量太大而不能穿过消化道上皮细胞之间的紧密连接),结果显示,除了peg2000-dspe之外的所有渗透促进剂均增加fd70转运量(图2g),这一结果与各吸收促进剂对上皮细胞紧密连接的破坏或上皮细胞的损伤一致。而在生理条件下,吸收促进剂可以增加10kda分子量的药物分子的转运,但不增加70kda分子量的分子的转运是理想的,因为这种尺度选择性会增强线性肽和小蛋白的递送,同时阻止分子量更大的内毒素的转运。
peg2000-dspe处理caco-2细胞2h可以导致通过细胞层的fd10转运增加2.1倍,但不允许fd70通过(图2e-g)。peg2000-dppe,peg2000-dope和peg1000-dspe对fd10的渗透都有类似的轻度影响,而peg5000-dspe则无此活性。综上所述,我们的数据表明并非所有两亲性聚合物都具有渗透增强剂的能力,但具有特定结构和化学组成的两亲性聚合物如peg2000-dspe在生物相容性好,无毒的基础上,具有高效且有选择地促进一定分子量范围内的大分子通过上皮细胞间隙进行转运的功能。当我们在caco-2或mdck单层上使用四种不同分子量的探针(荧光素、蓝色荧光标记的葡聚糖3kda、alexa647标记的fd10和tritc标记的fd70)重复同样的实验时,也获得了类似的结果(图3)。
实施例2、peg2000-dspe在正常生理范围内调节药物的细胞旁转运
peg2000-dspe有效地增强了fd10通过caco-2单层中的转运,同时不改变teer,不抑制细胞生长,这引起了我们的极大兴趣。物质通过不同的机制通过肠上皮细胞转运,主要包括细胞旁和跨细胞转运(13)。通过每个途径的吸收很大程度上取决于不同的物理特性,如分子量,亲水性和疏水性(15)。那么生物大分子如何以peg2000-dspe介导的渗透增强作用通过caco-2单层?为了回答这个问题,我们首先研究了在1mmpeg2000-dspe的存在下分子大小对通过caco-2单层的转运的影响,我们选择分子量范围为4至70kda的fd(葡聚糖)来比较它们的渗透行为。结果显示,
(1)在无peg2000-dspe存在的情况下,fd70几乎不能通过caco-2单层细胞发生跨细胞层渗透转运(图4a-b),fd20(papp=1.81×10-7cm/s)和fd40(papp=1.37×10-7cm/s)以几乎相同且非常低的渗透系数发生跨细胞层渗透转运,
(2)而在peg2000-dspe存在的情况下,fd20(papp=4.80×10-7cm/s)、fd40(papp=3.37×10-7cm/s)的渗透系数都显著增加。
(3)对于分子量更小的fd4和fd10,加入peg2000-dspe时,也显著增加了渗透系数,fd4渗透系数从papp=4.98×10-7cm/s增加至papp=9.83×10-7cm/s),fd10的渗透系数从papp=3.88×10-7cm/s增加至papp=8.56×10-7cm/s)(图4a-c)。
(4)与此相对的,0.5mmsds或2mmegta破坏了caco-2细胞层的形态完整性,并且导致2小时处理后caco-2细胞层对分子大小的依赖性限制渗透效应丧失(图4d-e)。
上述结果表明,peg2000-dspe的存在并未改变caco-2细胞层对分子大小依赖性限制渗透效应,仅增强了生理范围内caco-2单层的渗透性(及仅加速可通过caco-2细胞层的大分子的渗透速率,不能通过caco-2细胞层的大分子依然不能渗透通过)。
另外,peg2000-dspe对caco-2单层渗透的影响是暂时的和可逆的,在向caco-2单层添加
peg2000-dspe时,fd10的渗透系数快速增加至papp=11.5×10-7cm/s,而在洗去peg2000-dspe后,fd10的渗透系数立即恢复到基础水平papp=5.6×10-7cm/s(图4f-g)。
实施例3、peg2000-dspe选择性地调节zo-1分子的再分布
紧密连接(tjs)封闭上皮细胞的旁细胞间隙并产生细胞旁屏障,根据不同的情况,电导率,离子电荷偏好和不带电荷溶质的通透性水平不同(16)。为了研究peg2000-dspe如何作用于肠上皮细胞以及在生理范围内如何瞬时调节细胞旁途径,我们首先使用透射电子显微镜(tem)观察caco-2细胞单层和大鼠空肠处紧密连接在peg2000-dspe作用下的超微结构。所获得的tem图像显示,tjs位于caco-2细胞单层和大鼠肠上皮细胞的顶端侧,
(1)在对照组中,在tjs的位置处,膜紧密接近,在顶侧融合;
(2)用peg2000-dspe处理后,可以观察到tjs形态超结构的不同变化,例如细胞间隙的扩张或紧密连接周围的细胞质密度改变(图5a-b)。
越来越多的证据表明,tjs的结构处于动态调节的状态(17),渗透屏障的调控通常与tj蛋白的再分配有关(18)。随后我们用免疫荧光显微镜进一步分析peg2000-dspe对tjs蛋白,e-钙粘蛋白和f-肌动蛋白分布的影响。zo-1是tj蛋白之一,定位于紧密连接的细胞质表面,已被证实可与跨膜和细胞骨架蛋白相互作用,并参与细胞旁渗透通路的通透性调控(19,20)。
对caco-2细胞单层中的zo-1蛋白进行染色,显示了相邻细胞之间存在环状的连续结构,在用peg2000-dspe处理后,细胞-细胞接触区域的zo-1分散成片段化模式,并以颗粒结构发生位移进入细胞质(图5c)。
在peg2000-dspe处理后未观察到包括claudin-1,-2,-3,-4,-5,-7,-8,-10,-15和occludin在内的其他tjs蛋白的明显变化。此外,e-钙粘蛋白(e-cad)和f-肌动蛋白(actin)的免疫荧光染色也显示它们的定位和完整性未发生任何变化,表明在peg2000-dspe处理后对粘附连接和细胞骨架结构没有影响(图5c)。
为了研究peg2000-dspe改变tjs蛋白质的重新分布是否由于其影响tjs蛋白质的表达,我们用1mmpeg2000-dspe处理caco-2单层2小时(同时使用egta作为阳性对照),并分离成总细胞成分和胞质部分成分,然后western-blot标记zo-1,occludin和claudin-2。用zo-1,occludin和claudin-2抗体对这些组分进行免疫印迹可以了解peg2000-dspe处理后这些tjs蛋白的定位,这有助于进一步阐明peg2000-dspe对上皮tjs作用的模式。
结果显示,与对照相比,peg2000-dspe处理2小时后对整个细胞组分中zo-1,claudin-2和occludin的表达没有显示任何影响,同时,peg2000-dspe的处理并不诱导细胞质和膜之间的claudin-2和occludin的易位。但是,zo-1的胞质分布有明显的增加(142%),这意味着zo-1从紧密连接移动到胞浆(图5d)。相比之下,egta处理显著降低zo-1和claudin-2的总表达,但对occludin无影响,此外,egta处理导致zo-1,claudin-2和occludin都发生从膜转运至胞质的情况(图5d)。
以前的研究表明,tjs蛋白的动态行为的改变可能对渗透屏障的功能产生很大影响(21)。为了更直观的观察peg2000-dspe处理后活细胞中zo-1的动态行为的变化,我们构建了能表达mcherry-zo-1荧光蛋白caco-2细胞株,并对其进行实时成像观察。代表性细胞膜区域的不同时间成像结果显示了peg2000-dspe处理的细胞膜上zo-1的变化动力学,通过4小时的全细胞质膜运动追踪显示,peg2000-dspe的处理导致zo-1空间位置的变化和细胞膜形态的改变(图5e-f)。
综上,这些结果显示,尽管在peg2000-dspe处理过程中tjs蛋白的定位是动态改变的,但peg2000-dspe处理不直接影响caco-2细胞中tjs蛋白的总表达。通过peg2000-dspe处理改变上皮tjs的渗透屏障功能很可能仅与tjs结构的动态/物理改变相关。
实施例4、peg2000-dspe阻断紧密连接蛋白claudin-2的胞外环状结构域的相互作用
紧密连接蛋白claudin-2是小肠上皮细胞(例如caco-2细胞)中的主要内在膜蛋白(22)。claudins的拓扑结构具有四个跨膜片段、一个大的胞外环状结构域(eld1)和一个较短的胞外环状结构域(eld2,氨基酸序列为:wnlhgilrdfysplvpdsmkfeigek)共同组成。claudins能够通过粘合和聚合形成线性聚合物(称为紧密连接线),从而连接相邻的细胞并形成tjs的结构骨架(23-25)。为了研究peg2000-dspe是否重排tjs蛋白复合物,我们首先构建了基于脂质体的模型以通过claudins的胞外环域模拟相邻细胞之间的物理相互作用。合成c末端和n末端均被棕榈酰化的claudin-2的eld2结构肽段(pal-eld2s),然后将这些pal-eld2s分别插入由dio或dii标记的脂质体的脂质双层中,将dio标记的脂质体和dii标记的脂质体或pal-eld2修饰的这两种脂质体稀释并且盖在玻璃盖玻片上,并且通过共焦显微镜观察粘附在表面上的脂质体。未修饰pal-eld2s的脂质体表现为较小的红色或绿色斑点(图6a,左图),而pal-eld2修饰的脂质体显示出大的黄色斑点,其中两种染料共定位,表明不同颜色的脂质体发生相互交联融合(图6a,中间)。而一旦添加peg2000-dspe,这些大的黄色斑点消失并变回较小的红色或绿色斑点(图6a,右)。在分光光度计读板器中测量405nm波长处的光吸收也获得了相似的结果(图6b)。
为了进一步了解peg2000-dspe和pal-eld2之间的相互作用,我们使用等温滴定量热法(itc)来检测它们的结合亲和力。与具有不同peg链长和含有不同脂肪酰(pe)疏水核的其他pegs-pes分子相比,peg2000-dspe与pal-eld2的亲和力最高(表1),另外,peg2000-dspe增加pal-eld2二级结构中的α螺旋(图6c)。由于peg2000-dspe分子在两个嵌段中间具有带负电荷的磷基团,这可能对增强药物分子的跨上皮细胞的转运起了重要作用。为了验证这一假设,我们合成了两种peg2000-dspe类似物:
(1)没有负电荷的peg2000-dsg;
(2)带正电荷氮的peg2000-l-dsg(图7)。
这两个分子也能形成与peg2000-dspe相似的胶束结构,但是在caco-2细胞单层中,相比于peg2000-dspe分子,渗透增强功能显著降低,表明peg2000-dspe的磷基团对渗透性增强是至关重要的(图6d)。
表1、通过itc测量具有不同的peg链长度(1000-5000)的peg-pe与不同种类的磷脂和pal-eld2之间的结合亲和力
综上,我们证实了peg2000-dspe能够诱导关键tjs蛋白的重排,并通过与claudin-2的胞外环状结构域直接相互作用来阻断细胞间连接的形成,从而可逆地调节tjs的细胞旁通透性。
实施例5、蛋白质和多肽药物以自组装的方式装载到peg2000-dspe胶束中
我们的前期工作已经证明阿霉素,长春瑞滨和拓扑替康等小分子抗癌药物可以自组装到基于peg2000-dspe的聚合物胶束中(26,27),并且在药物与聚合物的适当摩尔比下获得约100%的药物装载效率(9,10)。本研究中,我们选择了胰岛素,鲑降钙素(sct)和胰高血糖素样肽-1(glp-1)作为模型蛋白和多肽类药物,这些载药胶束通过我们以前报道的方法制备(9,10,cn104288758b)。tem图像显示空胶束和载药胶束均为直径约为20nm的球形胶束,但载药胶束的粒径分布比空胶束更均匀(图8a-c)。
对装载效率的检测结果显示,在peg2000-dspe与胰岛素、sct和glp-1分别在5:1、3:1和2:1的摩尔比时,实现约100%的载药包封效率(图8d-f),通过计算itc(等温滴定量热法)的结合数也获得了类似的结果(图8g-i、表2),对于所有三种药物,滴定过程都是放热的(△h<0)和自发的(△g<0),表明这些药物可以自发加载到存在于水环境中的peg2000-dspe胶束中。所有三条滴定曲线看起来都是反向的s曲线,表明peg2000-dspe与药物之间存在典型的单一结合位点相互作用。
表2、itc方法检测到313k时peg2000-dspe胶束与三种药物相互作用的热力学参数
na:一分子药物平均结合的载体分子数量
我们通过超速离心来分析测定胶束装载后的胰岛素的状态,结果显示,在胶束中胰岛素主要是单体形式,由分子量为5,700da的峰表示(图9b),而在pbs溶液中的胰岛素主要以二聚体和六聚体形式存在,由分子量为12,000da和32,000da的两个峰表示(图9a)。此外,圆二色谱的检测结果也表明,对胶束装载胰岛素的圆二色谱(cd)谱图与胰岛素在ph1.4溶液中的cd谱图相似,但不同于ph7.4pbs溶液中胰岛素的cd谱图,也显示了胶束载体中胰岛素仅以单体形式存在(图9c-d和表3),这些数据进一步验证了包封在胶束中的胰岛素是以单体形式存在的。同时,装载在胶束中后,sct和glp-1的二级结构中的α螺旋构象变的更多(图9e-f和表4)。
表3、胰岛素在不同ph溶液中在m-ins中的圆二色谱数据
表4、装载在peg2000-dspe胶束中的三种药物的二级结构的变化
实施例6、peg2000-dspe增强药物的在体内消化道粘液层的扩散和跨肠上皮细胞层的渗透
蛋白质和多肽药物口服递送的第一个障碍是粘液层(30),peg2000-dspe胶束的亲水冠可减少胶束与粘蛋白的缔合并增加它们在粘液中的扩散。为研究这一问题,我们通过荧光漂白后恢复技术(frap)定量装载胰岛素(m-ins)的胶束在新鲜大鼠肠粘液中的粘液扩散参数,并以游离形式的胰岛素(f-ins)和装载胰岛素的脂质体(l-ins)作为对照。结果显示,m-ins形式下的fitc标记的胰岛素具有较快的移动性(t1/2为33s)和更多的可以快速移动的药物分子(60%),而相比之下,f-ins和l-ins形式的胰岛素因广泛地粘附于大鼠肠粘液层中而显示为更加固定的,l-ins的t1/2为133s,可移动药物分子比例为37%,对于f-ins,t1/2为80s,可移动的药物分子比例为43%,(图10a-b),表明m-ins具有比f-ins和l-ins更高的透过粘液层的能力。
蛋白质和多肽药物口服递送的第二大屏障是肠上皮细胞,因为不容易通过小肠上皮细胞的脂双层细胞膜扩散到血流中(31)。实施例2的结果显示了peg2000-dspe聚合物在caco-2细胞层中瞬时增强了fd10渗透性,为了进一步验证由通过与peg2000-dspe聚合物形成的胶束是否可以促进胰岛素跨过肠上皮细胞屏障,我们将装载的胰岛素胶束加入到caco-2单细胞层中,结果如图10c所示,通过caco-2细胞单层运输的m-ins形式的胰岛素的两小时累积量(47.6μg)远高于f-ins形式(21.8μg)。并且m-ins和f-ins的papp值分别为1.24×10-7cm/s和0.62×10-7cm/s(图10c)。进一步的,用荧光标记的m-ins(tritc标记的胰岛素和fitc标记的peg2000-dspe)孵育caco-2单层细胞并实时观察的结果显示荧光仅出现在细胞间隙中,这表明m-ins通过细胞间转运路线(图11)通过上皮细胞层。用pbs洗涤细胞后,仍能观察到来自fitc标记的peg2000-dspe的荧光位于细胞间隙,而未观察到来自tritc标记的胰岛素的荧光。这些结果进一步表明,peg2000-dspe聚合物通过从胶束中转移到细胞膜上,从而促进生物大分子通过细胞间隙转运。
为了进一步阐明peg2000-dspe胶束装载的胰岛素通过肠上皮细胞的转运机制,我们通过clsm观察穿过caco-2细胞单层的tritc-胰岛素转运方式。结果如图10d所示,在单独用tritc-胰岛素溶液孵育120分钟后,在5μm深度内的顶端侧仅检测到较弱的红色荧光信号。相比之下,当用荧光标记的m-ins时(tritc标记的胰岛素装载在fitc标记的peg2000-dspe胶束中)处理caco-2细胞单层时,在10μm深度内的顶侧观察到代表绿色和红色信号共定位结果的许多黄色斑点,显示m-ins胶束的结构依然是完好的。但是,在接近基底侧的较深扫描中只能看到红色信号,很难观察到绿色信号,表明胰岛素渗透穿过caco-2细胞层,但peg2000-dspe没有。这一结果表明,当胶束进入上皮细胞表面时,peg2000-dspe胶束将首先通过拆卸结构卸载其装载物,然后载体分子插入到细胞膜中的同时促进其装载物的转运。这种机制可能涉及peg2000-dspe胶束负载增加通过上皮细胞间隙的胰岛素渗透。
接下来,我们使用clsm观察体内大鼠小肠上皮细胞上fitc标记的胰岛素转运。如图10e所示,发现在pbs中溶解的fitc-胰岛素被阻碍在空肠粘液层的外部,这可能是由于胰岛素被高粘性粘液困住。相反,peg2000-dspe胶束中的fitc-胰岛素能够通过粘液并渗透进入空肠中的上皮表面,这将导致胰岛素通过肠上皮细胞吸收并进入全身血液循环,如在肠绒毛基底外侧和空肠肠系膜中所示(图10f)。针对胰岛素的免疫组化结果进一步证实m-ins中的胰岛素在2小时后定位于十二指肠空肠中,但pbs溶液中的胰岛素(f-ins)则没有在空肠中出现(图12)。正如我们所预料的那样,通过fitc标记的胰岛素(绿色)和alexa660标记的peg2000-dspe(红色)的共定位结果,反映出m-ins在空肠的粘液层中能够保持完整形式;观察到更大量的fitc-胰岛素位于肠绒毛的乳头侧,且其中绿色和红色的共同定位较少,表明m-ins胶束拆卸并卸载了装载的药物(图10g)。
综上所述,我们的研究结果证明了peg2000-dspe胶束可以通过粘膜层将它们装载的药物(例如胰岛素)携带至肠上皮细胞间隙,通过插入肠上皮细胞卸载药物并帮助药物穿过上皮细胞进入血液循环。
实施例7、在peg2000-dspe促进药物的跨肠上皮吸收,改善了药物代谢和药效
鉴于peg2000-dspe胶束装载的胰岛素表现出较高的肠上皮细胞透过性,然后我们考虑检查胶束中胰岛素的生物活性。对具有胰岛素依赖性(i型)糖尿病的大鼠测试了降血糖作用。正如预期的那样,对于皮下注射f-ins的大鼠,血糖水平在0.5小时内急剧降低,然后随时间增加(图13a),而在十二指肠给予f-ins后没有观察到降血糖作用,表明在缺乏合适的递送系统时口服胰岛素吸收不佳(图13b);同时,m-ins的十二指肠给药产生了显着的降血糖反应,在2小时内血糖水平降低最高达62.4%,并且在4小时内维持在大致相同的水平(图13a),该结果清楚地证明了口服m-ins能够有效降低糖尿病大鼠的血糖水平。图13b显示了相应的血浆胰岛素浓度-时间曲线,与f-ins十二指肠给药后几乎检测不到血浆胰岛素相比,m-ins十二指肠给药后,能够检测更高的血浆胰岛素浓度;用f-ins皮下注射的大鼠在给药后0.5小时产生最大胰岛素血浆浓度,而十二指肠给药的m-ins在2小时后显示了最大血浆浓度。通过计算m-ins(100u/kg)的十二指肠的auc(0-6h)与皮下注射胰岛素(5u/kg)的auc(0-6h)的比率,m-ins的相对生物利用度为4.63%(表5)。
同时另一蛋白质与多肽药物的模型-鲑降钙素的实验也进一步证明peg2000-dspe装载药物分子后可以促进药物的小肠吸收,发挥更好的药效学。如图13c所示,相比十二指肠给予f-sct组大鼠,m-sct组大鼠的降血钙更显著,6小时的血钙水平降低了40%,而f-sct组仅降低了约20%。图13d显示的血浆鲑降钙素水平结果表明,十二指肠给药m-sct在2小时后达到最大血浆浓度,其值显著高于f-sct组。通过计算m-sct(3.0mg/kg)的十二指肠的auc(0-6h)与皮下注射鲑降钙素(0.15mg/kg)的auc的比率,m-sct的相对生物利用率为9.6%(表6)。
表5、m-ins或f-ins通过十二指肠注射给药及f-ins通过皮下注射(sc)给药后的糖尿病大鼠(动物数量n=4)中胰岛素的药代动力学参数.
auc:血浆浓度-时间曲线下的面积;fr%:相对生物利用度(n=4)。
表6、m-sct或f-sct通过十二指肠注射给药及f-sct通过皮下注射(sc)给药后的正常大鼠(动物数量n=4)中鲑降钙素的药代动力学参数.
auc:血浆浓度-时间曲线下的面积;fr%:相对生物利用度(n=4)。
最后需要说明的是,以上实施例仅帮助本领域技术人员理解本发明的实质,不用做对本发明保护范围的限定。
sequencelisting
<110>中国科学院生物物理研究所
<120>一种促进胰岛素等大分子药物的口服吸收的吸收促进剂
<160>3
<170>patentinversion3.5
<210>1
<211>26
<212>prt
<213>homosapiens
<400>1
trpasnleuhisglyileleuargaspphetyrserproleuvalpro
151015
aspsermetlysphegluileglyglulys
2025
<210>2
<211>32
<212>prt
<213>人工序列
<400>2
cysserasnleuserthrcysvalleuglylysleuserglngluleu
151015
hislysleuglnthrtyrproargthrasnthrglyserglythrpro
202530
<210>3
<211>37
<212>prt
<213>人工序列
<400>3
hisaspgluphegluarghisalagluglythrphethrseraspval
151015
sersertyrleugluglyglnalaalalysglupheilealatrpleu
202530
vallysglyarggly
35
- 还没有人留言评论。精彩留言会获得点赞!