一种基于高通量计算筛选金属有机骨架催化材料的方法与流程
本发明属于计算化学与纳米复合催化材料领域,具体涉及一种基于高通量计算筛选高性能金属有机骨架催化材料的方法。
背景技术:
随着科技的不断进步与发展,人类的能源消耗量与日俱增,能源危机已经越来越受到人们的重视。我国则由于人口众多,人均能源储备较少而形势较为严峻,因此开发新型催化材料提升能源转换效率与能源利用率则显得尤为重要,金属有机骨架材料由于其独有的结构优势:例如高比表面积,规整的孔道结构,较多的活性位点等,目前在催化领域里已经得到了广泛的应用,然而目前开发金属有机骨架催化材料依然停留在“试错”的研究模式,这不可避免了带来了能源与资源上的浪费,而基于量子化学原理的高通量理论计算可以实现“计算先行,理论计算预测材料性能”,从而为材料的研究开发提供了一定的导向性,避免了盲目的试错从而极大的缩短研究周期,同时有效地解决了上述问题。
金属有机材料(mofs)是近年来发展极为迅猛的一种多孔配位聚合物材料,一般具有三维的孔道结构,以中心金属离子为节点,以有机配体支撑形成的网状延伸结构。目前mofs已经在催化领域里实现了广泛的应用。该类材料的比表面积远远大于有着相似孔道结构的分子筛材料,即使对材料进行多次填充也不会影响其孔道结构,且结构稳定性良好。而mofs中的中心金属离子,则因为其结构优势因而天然具备“单分散”的优点,非常适合作为催化材料的活性位点,同时,mofs结构对中心离子并不敏感,同一类型的mofs可以分别采用不同的中心金属离子,这大大扩展了mofs的应用范围,通过调节中心金属离子的类型可以满足多种催化反应的要求。
材料领域的理论计算大多基于密度泛函理论进行,它在量子力学的基础上实现了一次大的提升,伴随着现代高性能计算机技术的普及,使得高精度高通量的理论计算变得可行,理论计算也从最初的定性判断到了现在的定量解析的程度。目前高通量计算已经在催化领域里广泛采用,尤其是在催化机理分析,材料结构解析中发挥着越来越重要的作用,但是当前的理论计算大多数在材料合成后进行,主要运用在反应机理阐述中,未能充分发挥理论计算的指导性功能,因此开发一种高通量计算筛选mofs催化材料的方法可以有效地提升mofs的实用价值,缩短研发周期,有效地避免当前“试错”研究模式中的能源与资源浪费,具备广阔的实用前景。
技术实现要素:
本发明的目的在于通过高通量理论计算筛选高性能mofs催化材料,从而按催化反应的需求有选择地指导mofs的合成,使得mofs材料的研发具备了导向性,避免了传统“试错”的研究方法中的一系列问题。
本发明的技术方案是:首先针对待研究mofs系列与特定的催化反应体系,采用高通量并行计算的方式分别计算催化反应体系的吸附能,吉布斯函数变,活化能等一系列性能参数,从中选取性能最优的mofs材料。其次根据高通量计算结果,对最优mofs材料进行合成与表征验证性能,进一步反馈给相应的数据库,对高通量计算模式进行逐步的优化与完善。
具体操作步骤为:
(1)mofs材料的高通量计算筛选:
将待研究的mofs系列全部建模或者从数据库中直接抽提相关数据作为初始的输入文件。然后分别在同一水平下做相同的性质计算,具体的参数要求为:针对晶体结构优化采用gga-rpbe交换关联泛函,针对能带结构以及能态密度计算采用hse06屏蔽杂化泛函,所有计算默认的价层电子排布结构,赝势则采用ultrasoft赝势,截断能范围统一为550~800ev之间,monkhorst-pack格点取样采用高精度取样,scf自洽反应收敛阈值为5.0×10-7~1.0×10-5ev/atom,结构优化的收敛标准为能量波动范围小于5.0×10-6~1.0×10-5ev/atom,最大受力范围小于
(2)mofs材料的合成与性能验证及数据反馈
针对上述高通量计算结果筛选出的mofs催化材料,选取相应的可溶性金属盐与有机配体进行合成,合成方法包括但不限于水热法,溶剂热法,界面限制效应定向生长法,超声剥离法等等。在材料制备的基础上进行一系列表征与性能测试,并在实用化条件下进行实用化测试。通过实验验证将实验数据不断积累并进一步反馈给相应的数据库并对高通量计算模式进行逐步的优化与完善,不断提高计算效能。
进一步地,上述待研究的mofs包含了多种可溶性金属盐与有机配体合成的mofs:
进一步地,所述的待研究的mofs可用的可溶性金属盐包括:钛、钒、铬、锰、镍、铁、钴、铜、锌、锆、铝等金属的硝酸盐,氯化物,硫酸盐,醋酸盐等其中的一种或几种。
进一步地,所述的待研究的mofs可用的有机配体包括:1,4-对苯二甲酸、1,2-邻苯二甲酸、1,3,5-均苯三甲酸,1,2,4,5-均苯四甲酸,苯六甲酸,2-磺酸基对苯二甲酸,2-硝基对苯二甲酸,2-氨基对苯二甲酸,1,1':4',1”-苯基-4,4”-二甲酸,1,1'-二苯基-4,4'-二甲酸,哌嗪,吡嗪,三乙烯二胺,4,4'-联吡啶,1,3-二(4-吡啶)丙烷等其中的的一种或几种。
进一步地,所述的数据库包括crystallographyopendatabase(cod),cambridgestructuraldatabase(csd)等国际大型开放型数据库。所述的计算性质包含:能带结构,核心电子光谱,能态密度,反应路径以及活化能,电子密度,电子局域函数,核磁共振谱,分子轨道,原子布局分析,声子色散谱等。
本发明的优点在于:1)采用高通量并行计算的方式,有效地缩短了计算时间。2)采用理论计算指导材料合成与制备,使得材料的合成与制备具备导向性,避免了传统“试错”的研究模式带来的资源以及能源上的浪费,降低了研发成本,缩短了研发周期。3)通过实验数据实时反馈给理论计算这种交互式模式,使得理论计算的性能与精度得以不断提升与优化,具备良好的实用价值。
附图说明
图1(a)为高通量计算设置的基本设置参数页面,
图1(b)为高通量计算的电子项设置页面,
图1(c)为高通量计算的性质参数设置页面;
图2(a)为高通量计算下指导合成的二维mofs(nife-umns)的sem照片,
图2(b)为高通量计算下指导合成的二维mofs(cofe-umns)的sem照片;
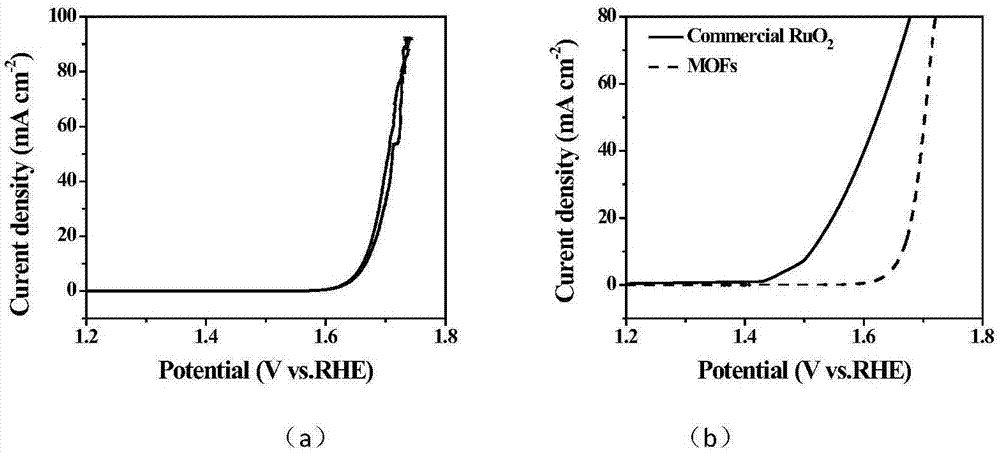
图3(a)为高通量计算指导下合成的nife-umns的电化学性能测试图谱,
图3(b)为高通量计算指导下合成的nife-umns的性能测试图谱;
图4为经过试验数据反馈后的计算结果,与实验数据符合的较为良好。
具体实施方式:
下面结合具体的实施方式对本发明的技术方案做进一步说明。
实施案例1
(1)对第四周期过渡金属离子与对苯二甲酸配体形成的mofs结构进行高通量计算筛选,晶格结构一律采用剑桥大学晶体结构数据库no.985792型晶体结构,通过组建不同中心金属离子的模型作为高通量计算的输入文件,该实施案例针对电化学催化体系,所有的计算均在同一计算精度下完成,结构优化时采用gga-rpbe交换关联泛函,针对能态密度计算采用hse06屏蔽杂化泛函,所有计算默认的价层电子排布结构,赝势则采用ultrasoft赝势,截断能统一为520ev,monkhorst-pack格点取样采用高精度取样(6×4×1),scf自洽反应收敛阈值为5.0×10-7ev/atom,结构优化的收敛标准为能量波动小于5.0×10-6ev/atom,最大受力小于
在计算指导下合成了nife-umns-mofs,其扫描电子显微镜照片如图2a所示,电化学催化活性性能如图3a,3b所示,经过试验数据反馈,进一步提升了计算的精度,如图4所示,与实验结果符合的较为良好。
实施案例2
对第四周期过渡金属离子与2-氨基对苯二甲酸配体形成的mofs结构进行高通量计算筛选,晶格结构一律采用剑桥大学晶体结构数据库no.985792型晶体结构进行修饰,通过组建不同中心金属离子的模型作为高通量计算的输入文件,该实施案例针对光化学催化体系,所有的计算均在同一计算精度下完成,结构优化时采用gga-pbe交换关联泛函,针对能带结构以及能态密度计算采用hse06屏蔽杂化泛函,所有计算默认的价层电子排布结构,赝势则采用ultrasoft赝势,截断能统一为580ev,monkhorst-pack格点取样采用高精度取样(6×4×1),scf自洽反应收敛阈值为5.0×10-7ev/atom,结构优化的收敛标准为能量波动小于5.0×10-6ev/atom,最大受力小于
- 还没有人留言评论。精彩留言会获得点赞!