用于生物路径中的调控互动的学习和识别的系统和方法与流程
相关申请的交叉引用
本申请请求共同待决的2012年10月9日提交的序列号为61/711491的美国临时专利申请、2012的年11月26日提交的序列号为61/729958的美国临时专利申请、以及2013的年1月18日提交的序列号为61/754175的美国临时专利申请的优先权。
本发明的领域是组学数据的计算分析,具体地本发明涉及针对路径分析的学习算法以及用途。
背景技术:
随着高速的遗传病筛查的出现,已经获得了捕获细胞的分子状态的逐步更大的数据集,而且这些进步实现了对癌症中改变的细胞机理的增强的识别和理解。例如,对特定肿瘤内频繁变化的关键目标的识别导致了最近20年来多达40种射靶疗法的开发。可惜的是,在大部分情况下,这些药物中的许多的反应速率低于50%,突出了这些药物所影响的路径的不完全理解。抗性机制的典型示例是结肠癌肿瘤中变化的egfr中的ras路径的激活,其中突变的kras构成地激活ras串,提供与egfr路径独立的生长信号,导致诸如西妥昔单抗治疗之类的egfr封闭疗法极大地无效。因此,看起来,西妥昔单抗对路径的干扰的知识相对于致瘤信号在细胞信令网络行进所经由的关键途径是不完整的。
由于用于在路径水平下对组学数据进行积分的大量计算工具现在变得可用,所以这种明显的不完整知识将更使人烦恼的。在各种其它工具中,多种算法(例如,gsea、spia和通路ologist)能够利用从文献编组的路径成功地识别出感兴趣的变化的路径。其它一些工具已经从文献中的编组互动创建了因果图,而且已经使用这些图来解释表达谱。诸如aracne、mindy和conexic之类的算法采用了基因转录信息(以及拷贝数,在conexic的情况下),由此很可能在一组癌症样本上识别出转录驱动器。然而,这些工具不会将不同驱动器组成识别单个关注目标的功能网络。诸如netbox和mutualexclusivitymodulesincancer(memo)之类的一些更新的路径算法试图解决癌症中的数据积分的问题,从而识别对样本的致瘤潜能关键的多种数据类型的网络。虽然这种工具允许路径上的至少一些有限积分以便找到网络,它们一般不能提供调控信息、以及该信息与相关路径或路径的网络中的一个或多个效应的关联。类似地,giena寻找单个生物路径中的解除管制的基金互动,但是不考虑路径的拓扑或者关于互动的方向或本质的先验知识。
外部遗传分析、概率图模型已经被广泛地用在网络分析中,其中具有贝叶斯网络和马尔可夫随机场形式的界标使用。多种方法已经通过多种不同方式从数据中成功地学习了互动,包括相关网络。更近时间以来,paradigm(在染色体组模型使用数据积分的路径识别算法)是在wo2011/139345和wo/2013/062505中描述了的一种染色体组分析工具,其使用概率图模型来在编组路径数据库上整合多种染色体组数据类型。这种模型系统有利地使得各个样本能够被单独访问或者在成群关注的环境下被访问。然而,在该工具中学习的期望最大化(em)参数仅仅缺省地在观察的数据参数上执行,因为有限尺寸的可用数据集抑制了互动参数的稳健评估。所以,该工具不能实现对将影响特定路径段中的活动的多种因素的互动和相互关系的分析,而且由此不能提供对流经细胞信令网络的信号流的改进的分辨率。
因此,即使学习和识别生物路径中的调控互动的大量系统和方法在本领域是已知的,所有或者几乎所有这些系统和方法具有一种或多种缺点。例如,在此以前已知的解析工具不能识别出调制路径的通路中的活动的参数的互动的强度和方向,由此不仅不能实现信号流和/或路径活动的干扰的预测,而且不能识别参数或路径元素的潜在差分使用。从不同观点来看,当前已知的工具通常仅仅考虑单独的基因活动,但是不能检查与调控链接相关的统计结果,因此仅仅提供了静态模型而不是动态模型。由此,已知的模型还不能检查网络中不同的调节器如何产生类似的细胞表型,尽管采用了完全不同的路径来实现它们。因此,仍然需要改进的系统和方法来学习和识别生物路径中的调控互动。
技术实现要素:
本发明涉及各种系统和方法,用于利用概率图模型来学习和识别生物路径中的调控互动,在概率图模型中路径模型具有经由各个通路而彼此耦接的多个路径元素。所提出的系统和方法中的通路被表达为具有根据多个互相联系的调控参数控制通路活动的调控节点。
根据组学数据集合和/或路径模型推断调控参数中的互动关联。由此,识别的互动关联现在能够识别出调整路径的通路中的活动的参数的互动的强度和方向。由此,设想的系统和方法能够预测路径活动的信号流和/或干扰,以及参数或路径元素的潜在的差别应用。从不同的观点来看,设想的系统和方法提供了动态路径模型,该动态路径模型可被用来识别通过一个或多个路径的信号流(甚至差分信号流)以及在各种(实际或仿真)情况下预测信号流。
在本发明主题的一个方面中,学习引擎包括组学输入接口,其接收一个或多个组学数据集合(例如,整个基因组数据、部分基因组数据、或差分序列对象)。组学处理模块耦接至该接口而且被配置成:(a)访问具有多个路径元素(例如dna序列、rna序列、蛋白质和蛋白质功能)的路径模型,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动,(b)经由组学输入接口获取组学数据集合中的至少一个组学数据集合,(c)基于所述至少一个组学数据集合和路径模型,推断多个调控参数中的一组互动关联,以及(d)基于互动关联来更新路径模型。更典型地,学习引擎还包括或耦接至染色体组数据库、bam服务器或排序装置。
在一些实施例中,路径元素包括dna序列,而且调控参数是转录因子、转录激活因子、rna聚合酶亚基、顺式调控元素、反式调控元素、乙酰化组蛋白、甲基化组蛋白和/或阻遏物。在其它实施例中,路径元素包括rna序列,而且调控参数是起始因子、转译因子、rna结合蛋白、核糖体蛋白质、小片段干扰rna和/或聚腺苷酸a结合蛋白,而且在另一些实施例中,路径元素包括蛋白质,调控参数是磷酸化作用、酰化作用、溶蛋白性裂解和与至少第二类蛋白的关联。
在特别优选的方面中,组学处理模块被配置成利用概率模型推断互动关联,概率模型使用共存和/或独立调控模型。而且,一般优选的是,概率模型进一步确定多个调控参数与通路的活动之间的依存关系的重要性和/或给出通路的活动的调控参数之间的条件依存关系的重要性。此外,设想的是,概率模型进一步确定调控参数的互动的符号。
因此,从不同的观点来看,发明人还设想一种产生路径模型的方法,其包括经由组学输入接口获取至少一个组学数据集合(例如,整个基因组数据、部分基因组数据、或差分序列对象)的步骤。设想的方法还包括经由组学处理模块访问具有多个路径元素的路径模型的附加步骤,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动;以及基于至少一个组学数据集合和路径模型,经由组学处理模块推断多个调控参数中的一组互动关联的附加步骤。在另一步骤中,基于互动关联来更新路径模型。更典型地,从染色体组数据库、bam服务器或排序装置获取组学数据集合。
在本发明主题的另一方面中,推断步骤基于概率模型,而且最优选的是,概率模型使用共存和/或独立调控模型。此外,设想的方法包括确定多个调控参数与通路的活动之间的依存关系的重要性和/或给出通路的活动的调控参数之间的条件依存关系的重要性的步骤。还优选的是,设想该方法包括确定调控参数的互动的符号的步骤。
在本发明主题的另一方面中,一种用于针对路径模型中的调控节点的调控参数识别出亚型特定互动关联的方法,包括:经由组学输入接口获取表示亚型组织的至少一个组学数据集合的步骤,以及经由组学处理模块访问具有多个路径元素的路径模型的步骤,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动。设想的方法还包括:经由组学处理模块通过多个调控参数间的互动的概率分析从表示亚型组织的至少一个组学数据集合导出亚型互动关联的步骤,以及在路径模型中呈现导出的亚型互动关联的另一步骤。在特别优选的方面中,亚型组织是抗药性组织、分生组织、药物治疗组织或组织的克隆变异体。
在需要时,设想的方法还可包括利用生物体外、硅中和整体实验中的至少一个来验证导出的亚型互动关联的步骤。
在本发明主题的另一方面中,发明人设想了一种将表示组织的组学数据集合归类为属于亚型特定组织的方法。该方法典型地包括a经由组学输入接口获取表示组织的组学数据集合的步骤,以及针对组学数据集合导出路径模型中的调控节点的多个调控参数中的一组互动关联的另一步骤。在另一步骤中,导出的一组互动关联与和已知亚型特定组织相关的先验已知的一组互动关联进行匹配,而且该匹配被随后用来归类出表示组织的组学数据集合属于已知亚型特定组织。
更优选地,获取步骤包括从具有已知调控特征的组织的组织样本(例如,肿瘤样本)产生表示组织的组学数据集合,而且已知亚型特定组织是抗药性组织、分生组织、药物治疗组织或组织的克隆变异体。
在本发明主题的另一方面中,发明人设想了一种识别具有多个路径元素的路径模型中的药物靶点的方法,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动。该方法包括下述步骤:(a)经由组学输入接口获取表示组织的组学数据集合,(b)针对组学数据集合导出路径模型中的调控节点的多个调控参数中的一组互动关联,以及(c)在预测药物干扰了互动关联处将药物识别为影响了通路的活动。更典型地,调控节点影响了蛋白质的转录、转译和后转译改型中的至少一个,而且药物是商用药物而且具有已知模式的作用。
在本发明主题的另一方面中,发明人设想了一种识别具有多个路径元素的路径模型中的目标路径的方法,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动。该方法优选地包括经由组学输入接口获取表示组织的组学数据集合的步骤,针对组学数据集合导出路径模型中的调控节点的多个调控参数中的一组互动关联的另一步骤,以及根据药物对互动关联的已知效果将路径识别为目标路径的又一步骤。
更优选地,已知效果是对致活酶的抑制效果、对受体的抑制效果、以及对转录的抑制效果中的至少一个。在其它适当目标路径中,尤其设想的目标路径包括钙/钙调蛋白调控路径、细胞激素路径、趋化因子路径、增长因子调控路径、荷尔蒙调控路径、map致活酶调控路径、磷酸酶调控路径和ras调控路径。该还可包括根据识别出的路径提供治疗建议的步骤。
因此,设想的方法还包括一种经由电脑模拟仿真药物的治疗效果的方法,包括:获取具有多个路径元素的路径模型的步骤,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动。设想的方法还包括识别出对至少一个调控参数的影响已知的药物的步骤;通过组学处理模块并基于药物的已知效果,经由电脑模拟而改变路径模型中的调控节点、活动和至少调控参数中的至少一个的另一步骤;以及确定路径模型中的改变的次生效应的又一步骤。更典型地,次生效应在路径模型中的另一调控节点、另一活动和另一调控参数中。
本发明主题的各种目标、特征、方面和优势将由优选实施例的下述详细说明以及附图而变得更明显,附图中类似标号表示类似元素。
附图说明
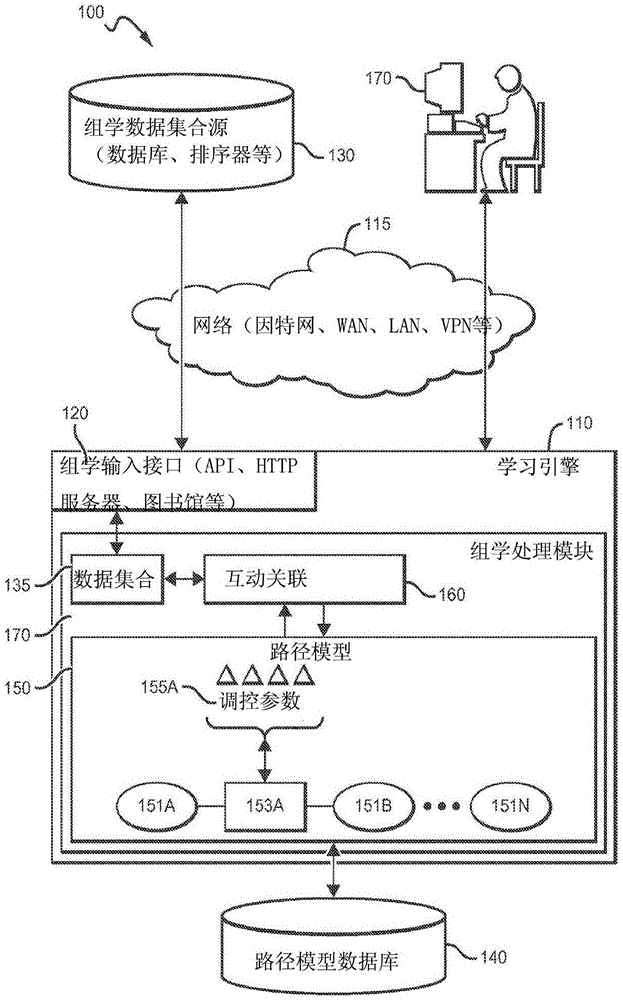
图1是根据本发明主题的学习引擎的示例性示意表示。
图2a是根据本发明主题的因子图结构的示例性示意表示,而且图2b示意性地描绘了针对转录、转译和激活节点的替换调控模型。
图3a是针对整个tcga群中学习的每个互动的wpmi矢量的主成分分析(pca)的示例性示图。图3b图示了路径中的标记为激活和抑制的明显链接的群集成员,而且图3c示出了群集的几何中心的wpmi值的热点图,示出了从最强抑制至最强激活的范围。
图4a和4b是信息化(4a)和单调(4b)初始化下的明显链接的wpmi值的群集成员条形图。
图5a是描绘了未通过学习完整的条件概率的循环的每个em步骤下的后续测试的唯一子节点的百分比的示例性示图,而且图5b是连贯对照不连贯三联体的示例的示意图示。
图6a-6c是描绘了针对采用不同解析方法的路径分析的kaplan–meier存活曲线的示例性示图。
图7是g-分排名的示例性热点图表示g-分排名。
图8a-8b是描绘了用于激活来自ppara-rxra和tap73a的链接的组织所组合的wpmi信号的示例性盒型图。
具体实施方式
发明人现在已经发现,可以实现概率图形路径模型,其中调控参数的相互关系被统计确定。由此,设想的系统和方法的分析和仿真将提供明显改进的精确度,而且实现了不同路径和/或亚组织内的调控元素的差别使用的识别。
因此,应该注意的是,通过利用群中的关注表型内的显著不同的使用分布识别调控链接,就可能检查网络中的不同调节器是如何产生类似的细胞表型,哪怕使用完全不同的路径来实现它们。此外,这样学习的参数可被用作统计测试的基础以针对每个调控节点建立同生群的各个样本或子集在多大程度上遵循之前学习的参数模式的分布。
在上述讨论通篇,大量参考考虑了服务器、服务、接口、门户、平台或由计算装置形成的其它系统。应该理解的是,这种术语的使用被认为代表了具有至少一个处理器的一个或多个计算装置,该至少一个处理器被配置用于执行存储在计算机可读的有形的非易失性介质上的软件指令。例如,服务器可包括一个或多个计算机,其以满足上述较色、责任或功能的方式操作作为网络服务器、数据库服务器、或其它类型的计算机服务器。
例如,图1示例性地描绘了生态系统100,其包括学习引擎110。学习引擎110被配置成针对一个或多个路径模型150处理一个或多个组学数据集合135。学习引擎110包括两个主要部件:组学接口120(学习引擎110经由组学接口120获取关注的数据集合)以及组学处理模块170(其被配置成分析数据集合)。在所示示例中,学习引擎110被图示为可通过网络115(例如,因特网、wan、lan、vpn、nationallambarail(参见urlwww.nlr.net),等)访问的计算装置,很可能是http服务器工厂。在一些实施例中,学习引擎110通过网络115提供收费服务。例如,学习引擎110可经由基于云的平台即服务(paas)、基础设施即服务(iaas)、软件即服务(saas)、或其它类型的服务暴露一个或多个组学输入接口120给分析员170或其它用户。在其它实施例中,学习引擎110可以是相对于分析员170的本地计算装置并且被配置成运行承担上述学习引擎110的角色和责任的一个或多个软件指令包。
组学输入接口120代表配置用于接收一个或多个组学数据集合135的计算接口。接口120的一个示例可包括能够经由网络115接收数据集合135的http服务器。例如,数据集合135可包括序列化格式(例如,xml)、bambam格式、或可通过http服务器传输的适当数字格式的文件。在其它实施例中,接口120可具有应用程序接口(api)的形式,数据结构或它们的参考可通过其而经由网络115传递至学习引擎110,作为远程程序调用或甚至经由本地库功能调用。应该理解的是,组学输入接口120可被配置成耦接至一个或多个组学数据集合源130,很可能操作作为数据库。在一些实施例中,学习引擎110包括与组学输入接口120耦接的染色体组数据库或排序装置。
组学数据集合135可包括较宽范围的组学数据。在更优选的实施例中,组学数据集合135表示染色体组数据,很可能是整个基因组数据、部分基因组数据、差分序列对象或其它染色体组数据。而且,组学数据集合135还可表示其它类型的数据,包括蛋白质、代谢组学、脂类组学、动力学、或其它组学数据形态。
处理模块170表示与组学输入接口120耦接的计算装置的至少一部分而且被配置成针对路径模型150分析数据集合135。处理模块170的一个方面包括访问一个或多个路径模型150的能力,可能地来自路径模型数据库140或其它模型源。在一些实施例中,组学处理模块170还可利用组学输入接口120来访问路径模型数据库140。
路径模型150表示将被建模的目标组学系统的活动的数字模型,可能地为因子图的形式。每个路径模型150包括多个路径元素151a至151n,统一地称为路径元素151。路径元素151表示沿通路的发生活动的级。在至少两个径元素151之间,例如所示的路径元素151a和151b,是调控节点153a表示的调控节点,总体上称为调控节点153。虽然未示出,但是路径元素151的每个集合之间可以存在其它调控节点153。因此,至少两个路径元素151,例如路径元素151a和151b,经由具有调控节点153(如所示的调控节点153a)的通路彼此耦接。路径模型150的调控节点153根据一个或多个调控参数155a(总体上称为调控参数155)控制元素之间的沿通路的活动。应该理解,路径模型150可包括任意可行数量的路径元素151、调控节点153和调控参数155。举例来说,考虑其中路径元素151包括dna序列、rna序列、蛋白质、蛋白质功能或其它活动元素的情况。
在其中路径元素151包括dna序列的情况下,调控参数155可包括转录因子、转录激活因子、rna聚合酶亚基、顺式调控元素、反式调控元素、乙酰化组蛋白、甲基化组蛋白、阻遏物或其他活动参数。此外,在其中一个路径元素151包括rna序列的情况下,调控参数155可包括起始因子、转译因子、rna结合蛋白、核糖体蛋白质、小片段干扰rna、聚腺苷酸a结合蛋白或其他rna活动参数。而且,在其中一个路径元素151包括蛋白质的情况下,调控参数155可包括磷酸化作用、酰化作用、溶蛋白性裂解和与至少第二类蛋白的关联。
组学处理模块170利用路径模型150以及数据集合135来推断多个调控参数中的一组互动关联160。可被用来推断互动关联160的一个示例类型模型包括概率模型,其中模型配置组学处理模型170以比较多个原始数据集合135的多对调节器参数。在一些实施例中,调节器节点153根据共存调控模型进行操作,其中学习引擎110学习给定父节点的子节点的完整的条件概率表。在其它情况下,调控节点153可基于独立调控模型进行操作,其中学习引擎110利用
设想的概率模型被进一步配置成确定多个调节器参数155之间的依赖性以及相应通路的活动的重要性,给出通路的活动的调控参数之间的条件依存关系的重要性。例如,一旦计算出或者建立了条件概率,组学处理模块150可利用g-测试来确定重要性。而且,概率模型被进一步配置成针对调控参数确定互动的符号。一旦建立了互动关联160,路径模型150可被更新来反映学习到的互相关系。由此,应该理解,学习引擎通常包括组学输入接口,其接收一个或多个组学数据集合。这种组学输入接口可被耦接至在大部分典型情况下向组学处理模块提供了组学信息的各种装置或系统。例如,组学信息可从公开数据、基因组的、rnomic和/或蛋白质组的组学数据库导出,来自组学信息数据库(例如,tcga)的输出文件,以及提供了组学数据的其它装置、服务和网络,包括dna、rna和/或蛋白质序列数据库,排序装置、bam服务器等;由此,应该理解的是,数据的格式可考虑变化而且可表示为整个基因组数据、部分基因组数据、或差分序列对象。
更典型地,组学处理模块在信息上耦接至接口而且被配置成访问具有多个路径元素(例如,dna序列、rna序列、蛋白质和蛋白质功能)的路径模型,其中两个或更多元素经由具有调控节点的通路彼此耦接,调控节点根据多个调控参数控制沿通路的活动,(b)经由组学输入接口获取至少一个组学数据集合,(c)根据至少一个组学数据集合和路径模型,推断多个调控参数中的一组互动关联,以及(d)根据互动关联更新路径模型。
应该认识到,对于路径模型:(a)可由组学数据的集合产生,或者可从之前的确定结果获取。因此,设想的系统和方法将包括耦接至组学处理模块的存储模块,其中存储模块存储一个或多个之前确定的路径模型。还应该认识到,存储的路径模型可对应于‘正常’组织或生病组织。当路径模型来自生病组织时,还应该认识到,生病组织可具有由亚特点表征的特定亚型(例如,对针对特定药物抗治疗的亚型,来自转移性组织的亚型,等等)。还设想,组学数据可按照各种方式经由接口提供。例如,数据可被提供为单个文件,或者不同的多个文件的集合,其可由服务提供商提供,来自之前存储的库,或来自排序装置或序列分析系统。因此,学习引擎可还包括或者可被耦接至染色体组数据库、bam服务器或排序装置。
根据具体通路,应该注意的是,路径元素的特性将显著变化,而且随着调控参数的特性变化。然而,总体上应该注意,调控参数将确定流经从路径元素至下游元素的通路的信号流。例如,在路径元素是或者包括dna序列时,设想的调控参数将是dna序列的影响转录(或其它任务)的那些细胞实体。因此,针对dna序列的设想的调控参数包括一个或多个转录因子、转录激活因子、rna聚合酶亚基、顺式调控元素、反式调控元素、(脱)乙酰化组蛋白、(脱)甲基化组蛋白和/或阻遏物。类似地,在路径元素是或者包括rna序列时,设想的是,适当的调控参数包括影响rna的转译(或其它活动)的因子。由此,这种调控参数包括起始因子、转译因子、rna结合蛋白、核糖体rna和/或蛋白质、小片段干扰rna和/或聚腺苷酸a结合蛋白。类似地,此处,路径元素是或者包括蛋白质,影响该蛋白质的活动的所有因素被认为是适当的调控参数而且可因此包括其它蛋白质(例如,与蛋白质互动以形成激活的复合体或与不同活动复合),化学改型(例如,磷酸化作用、酰化作用、溶蛋白性裂解等)。
相对于调控参数中的该组互动关联的推断,总体上设想,该推断基于组学数据集合和/或路径模型,而且总体上设想,利用下文将更详细描述的概率模型(例如,共存和/或独立调控模型)执行该推断。由于潜在的非常大量的可能互动关联,而且总体上设想,组学处理模块将确定(单个节点的)调控参数和通路的活动之间的依赖性的重要性水平和/或给出通路的活动的(单个节点的)调控参数之间的条件依存关系的重要性。按照这样的方式,解析焦点可被分配给具有统计上最高的重要性的互动关联,下文将更详细地描述。
未限制至本发明主题,发明人还发现,互动关联及其重要性的互动关联可进一步通过针对调控参数确定互动的符号(正/激活,或者负/抑制)的统计操作进行提炼。利用由此确定的互动关联及其对通路的影响现在将提供对路径的网络和通过该路径的信号流的显著改进的理解。
因此,从不同的观点来看,应该理解的是,可通过经由组学输入接口获取至少一个组学数据集合(例如,整个基因组数据、部分基因组数据、或差分序列对象)产生路径模型。组学处理模块随后访问具有多个路径元素的(例如,之前确定的)路径模型,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动。组学处理模块随后根据组学数据集合和/或路径模型推断多个调控参数中的一组互动关联,而且路径模型随后根据互动关联进行更新。
类似地,应该认识到,利用设想的系统和方法,可以识别出针对路径模型中的调控节点的调控参数的亚型特定互动关联。如上所述,经由组学输入接口获取表示亚型组织的至少一个组学数据集合,而且组学处理模块访问之前确定的路径模型。随后经由组学处理模块,通过多个调控参数中的互动的概率分析,从代表亚型组织的组学数据集合导出亚型互动关联,下文将更详细地描述,而且导出的亚型互动关联随后呈现在(或并入)路径模型中。虽然组织的所有类型的亚类型被认为对于此处的使用是适当的,特别设想的亚类型包括抗药性组织、分生组织、药物治疗组织或/或组织的克隆变异体。实验的和/或理论的实验(例如,生物体外、硅中、整体)可随后可被执行来验证导出的亚型互动关联。当然,相对于该方法的组件,与上述和下述相同的考虑可应用。
更具体地,在此处呈现的概率图模型中,来自样本(例如,肿瘤活体组织切片)的生物分子(例如蛋白质、信使rna、复合体和小生物分子)的状态在此表现为变量。例如,对于每个基因,变量被用于该基因的基因组副本数、转录该基因的信使rna、从该基因导出的蛋白质,而且在大部分情况下,对应于可被蛋白质的后转译改型调控的基因(在路径中注释)的生物活动的非物理附加变量。变量还可被包括来表示路径中一般注释的更多抽象状态,例如细胞死亡。
改变分子的状态(例如基因转录调控、蛋白质磷酸化作用、复合物形)的有原因的互动被表现为从调控变量至被调控变量的指向边缘。因此,对于模型的概率图中的每个变量y,因子被引入将变量状态关联至所有其调节器的状态的联合概率模型:f(y|x1,x2,…,xn),其中x1至xn是调控y的变量。该因子是条件概率表格:对于parents(y)的每个设定,σy∈f(y=y|parents(y))=1。各个变量的观察,例如基因组副本数或基因表达,被建模成单独的变量,通过因子f(y|x)连接至潜变量,也是条件概率表格。全联合概率状态则是:
其中z是路径中的调控循环所需的归一化常数。
给定对样本的观察,则可以利用具有概率空间中执行的推断(与空间相反)、10-9的收敛公差以及具有seqfix更新方案的libdai中的环环相扣的信念传播实现,解决每个未观察到的变量的临界分布。在机器学习处理中经由libdai中的期望最大化来学习针对所有f函数的参数,在连续自然对数可能性的比例小于10-10时停止。
应该理解的是,发明人现在已经引入新变量至与每个基因的转录、转译和蛋白质调控状态对应的每个基因的中心法则,如描绘典型因子图结构的图2a所示。该中心法则意味着,每个蛋白质-编码基因将具有相同的中心法则结构,而且因此可能在所有基因中共享参数。随后在每个基因的转录、转译和蛋白质调控变量中建模调控程序。
调控模型
之前开发的算法(如wo2013/062505和wo2011/139345中所述,它们在此通过引用而并入)通过改变调控节点如何被算法处理来扩展。为了构建因子图并且实现多类数据间的比较,之前开发的算法将输入数据离散化为相对于一些控制的上、下和正常。调控节点收集沿从dna至有效蛋白质的通路的一些点处的给定基因的调控中涉及的所有基因的活动信号。这些信号被收集在通过因子连接至基因的中心法则结构的单个变量中。在之前开发的算法下,调控节点简单地对输入信号表决以决定激活或抑制信号是否通过。
相反,在根据本发明主题的系统和方法中,利用机器学习处理来学习通过父节点x1,…,xn的给定设定的子节点变量y的每个设定的可能性。在下文中,共存和独立调控模型形成对照并示例地示出在图2b中,表示针对转录、转译和激活节点的替换调控模型。在共存调控模型中,给定父节点的子节点的完整的条件概率表格被学习,同时在独立调控模型中,各个链接的条件概率被学习,而且naivebayes假设被用于计算给定父节点的子节点的概率。
更具体地,利用共存调控模型,概率直接被存储为针对父节点和子节点的所有可能设定的统计概率表格中的参数。相反,利用独立调控模型,p(y)和p(xi|y)被用作参数而且参数的乘积被计算来得出下面的概率:
其中z是对应于p(x1,…,xn)的归一化常数。为了针对独立调控模型初始化参数,p(y)被分配下、上和正常的相当概率,而且基于路径中的链接的注释设置针对p(xi|y的初始概率)。对于在注释中标记为催化剂p(down|down)=p(normal|normal)=p(up|up)=0.8的链接,而且对于抑制剂p(down|up)=p(normal|normal)=p(up|down)=0.8,所有其它设置的所有概率被设置为0.1。利用所有设置上的均匀分布执行测试,以评估采用路径的先验知识的重要性。相同的简单表决程序随后被初始地用于之前开发的算法作为针对共存调控模型中的em学习的初始参数。在∈=0.001时,遵循99.9%的概率处于表决胜出的子节点状态中,而且0.05%处于其它状态中作为初始可行性。
此外,发明人还实现蛋白质和有效状态之间的复合体和基因系列的‘激活’调控。具体地,每个系列和复合体现在通过三个一组的变量进行建模:系列/复合体,调控和有效,与单个因子f(active|regulation,family|complex)连接。系列或复合体的调节器被连接至有效变量,共存或者独立调控模型。系列或复合体的组分利用噪声最小或噪声最大因子连接至系列/复合体变量,其中∈=0.001。相反,仅仅噪声最小或噪声最大因子被用于之前开发的算法中。
调控统计
发明人使用g-测试来确定调控链接(第一等式)的父节点和子节点之间的依赖性的统计重要性以及给定子节点的父节点分布(第二等式)之间的条件依赖性的统计重要性:
应该注意的是,g-测试遵循x2分布,由此可分别针对父节点–子节点测试和父节点-父节点测试利用4和12个自由度的x2分布得到p-值。针对错误发现率(fdr)调节p-值,而且调节成p<0.05的链接被着重考虑。虽然g-测试(其正比于共同信息)有益于知道互动有多强,其并没有提供互动符号的细节(激活是正互动,抑制是负互动)。
为了获取该信息,发明人计算了父节点和子节点之间的pearson关联以及加权的逐点共同信息,或者父节点和子节点的所有可能设置处的wpmi(参见下面的公式)。利用联合分布p(xi,y)=p(xi|y)p(y)计算关联,而且利用fisher转化计算重要性。给定子节点的父节点之间的关联也被计算来确定是否三个节点形成连贯或不连贯前馈环路。为了比较组之间的g-测试结果,采用每个组中的g统计的等级的差异。通过执行具有组成员的5000个随机排列的排列测验并随后调节fdr来计算该统计的重要性。对于大于排列中观察到的这些中的任意一个的差异,最低可能的p-值被用作上界。
因此,应该认识到,wpmi仅仅是g-分数和的每个单独的元素,而且9个wpmi值的矢量可被布置成容易解释热点图。数据可利用hopach群集算法(来自bioconductor)进行分析,其试图找到最好地符合数据的群集数。这就导致了针对ipl群集的每个集合的不同数量的群集。为了找到具有所有数据集合之间的一致数量的群集的群集,发明人通过将小群集成员重新分配给最近的大群集,从而瓦解了最小群集,而且按照这样的方式瓦解了小群集以得到所有数据集合之间的一致数量的群集。该方法还用于在我们比较期间保持群集尺寸恒定。
示例
存在大量方法来产生路径模型,代表模型由2012年2月27日格式的biopaxlevel3中下载的biocarta的reactome、pid和ncipid解析产生。该路径模型包括7111个蛋白质、52个rna基因、15个微rna基因、7813个复合体、1574个基因系列以及586个抽象生物过程。存在改变分子的激活状态的8603个互动(3266个抑制剂)、2120个转录激活链接以及397个转录阻遏链接,而且存在针对7813复合体的24129个组分以及针对1574个基因系列的7170个成员。
发明人使用david来执行对本发明系统和方法中学习的互动中涉及的基因的基因集合富化。为了最大化david识别出的基因数量,基因复合体和系列被分成它们的组分基因。针对链接中涉及的富化与编组路径中的所有基因的背景进行比较。
具有n个父节点的完整的条件概率表格将存储父节点和子节点的所有3n+1个可能设置的概率。由于编组路径中的一些中心基因具有多过30个的调节器,可附接至子节点的父节点的数量被限定至5个以防止这些表格的尺寸变得过高。对于多余5个蛋白质调控的基因,中间节点被添加至示图以保持该限定。因此,具有10个调节器的基因将具有两个中间节点,其中每个中间节点附接五个调节器。
使用具有来自11个组织类型的基因表达和副本数量据的1936个tcga肿瘤样本的数据集合,互动和调控互动被学习,互动重要性被g-测试确定,而且理由上述关联值确定互动符号。在调控蛋白质的路径模型中的9139个互动中,7631个(83.5%)被发现在0.05的fdr时明显。针对在整个tcga群上学习的每个互动的wpmi矢量的主成分分析(pca)揭示了从强禁止至强激活的梯度。示例性主成分分析在图3a-c中示出。此处,面板(a)图示出tcga群中的调控链接的主成分分析,其中每个点是9个wpmi分数针对链接在顶部的两个主要组分上的投影。凸包示出了(未投影)wpmi分数上执行的k平均聚类群集的成员,而且群集成员被布置在每个群集的几何中心。面板(b)图示了路径中标记为激活和抑制的明显链接的群集成员,面板(c)示出了群集的几何中心的wpmi值的热点图,示出了从强抑制(1)至强激活(5)的范围。沿该梯度的群集找到的wpmi矢量的k平均聚类群集表示规范互动类型从强激活至强抑制的范围。在7631个明显链接中,78个(1%)被布置在其中图心走向与路径中如何注释链接相反的方向的群集中。大量wpmi矢量示出了em能够学习具有相似的催化剂和抑制剂以及更复杂的调控模式的新互动制度。
使用统计关联测量(见上文),发明人随后访问每个互动(激活或抑制)而且与路径模型中注释的互动类型比较。存在具有显著关联和g-分数的7357个链接,而且这些中,219个链接(3%)的关联与路径中的调控的方向不一致。这利用了就测试和与编组链接一致而言显著的7138个(78%)链接。发明人还从g-测试发现一些链接具有高关联值但是具有低重要性,这在通常在其中父节点或子节点分布极大地有利于单个状态的情况下观察到。
在本发明方法学习的链接中,1197个具有显著关联和g-分数而且不包括包括复合体或系列。对于这些链接中的51个链接(4.3%),关联洗漱的符号与文献不一致。另一方面,仅仅看基因表达谱,1058个非复合体非系列链接被发现具有显著关联,但是470个(44%)与路径入口的符号不一致。对于第二比较,通过将系列和复合体的组分的所有基因直接连接至这些系列和复合体调控的任意基因来消除复合体和系列。该压扁程序导致了200921个链接。我们发现这些链接中的165258个显著地关联基因表达谱,而且链接中的81558个(49.4%)具有与路径中的链接的方向不一致的关联。这些结果表明,本发明方法学习的链接比基因表达谱的关联显著地更与文献中的链接的方向一致。
仅仅对从tcga卵巢癌症(ov)病人(n=416)学习的wpmi分数运行pca和群集分析而不进行复合体和系列激活调控,产生了与图3a和3c所示的pca和群集中心非常类似的结果,但是发现更少的明显链接和更高比例的注释为催化剂的链接,而且被学习为抑制剂或者反之亦然(图4a)。当p(xi|y)=1/3(图4b)的平的初始化被采用,发明人发现群集中心再次被映射至从激活至抑制的梯度,而且相对于包括方向信息的初始设置,存在更少的明显链接和更高比例的链接方向一致性。
为了测试图2中的naivebayes独立假设,根据本发明构思的系统和方法以对tcga卵巢癌症样本的独立和共存调控模型来运行。发明人对运行的每个em步骤计算的期望值测试了条件独立性假设(参见图5a)。图5a图示了未通过学习完整的条件概率的运行的每个em步骤处的后续测试的唯一子节点的百分比(文字说明:i.给定子节点的任意两个父节点的条件独立性的重要性的测试。ii.测试i和至少未通过的一个父节点被明显链接至子节点。iii.测试i和失败三联体是不连贯的。iv.测试i、ii和iii。在学习的每个步骤,更少的共存调节器被发现彼此独立。由于路径中的小反馈回路,例如调控其本身的转录的转录因子,可以预期独立性假设在某些情况下不成立。此外,很常见的是,仅仅一个系统不同的两个非常类似的复合体会共存调节相同子节点,该情况下还可预期条件测试不通过,哪怕存在小冲突。由此,发明人将其中其中两个共存调节器未通过独立性测试的情况划分为‘连贯’和‘不连贯’类别,如图5b所示。图5b图示出连贯对照不连贯三联体的示例。箭头对应于针对正关联(激活)的具有尖头的关联以及针对负关联(抑制)的平头。父节点之间的互动在文献中找到,由此双侧箭头被采用,因为该互动的方向是未知的。
此外,由于其它调节器的强度的原因,两个共存调节器可能不通过独立性测试,即使一个共存调节器是不明显调节器。发明人因此还考虑其中两个共存调节器自己是明显的情况的子集,而且测试显示加权表决方法产生的初始参数导致了几乎50%的子节点未通过调节独立性测试,但是随着em算法学习更可能的参数设置,越来越少的节点未通过测试。通过组合我们所有的测试,显示仅仅大约5%的子节点很可能以一种有意义的方式具有共存调节器。
利用卵巢癌症样本,发明人还群集了之前开发的算法(参见wo2013/062505和wo2011/139345)以及来自共存和独立调控模型两者的算法产生的蛋白质活动预测。随后对这些群集执行kaplan–meier分析以查看是否它们具有显著不同的存活情况(图6)。此处,利用之前开发的算法示出了综合路径活动所群集的tcga卵巢群中的416个病人中的kaplan–meier存活曲线(图6a),(图6b)本发明算法学习调控节点的完整的条件概率表格,而且(图6c)本发明算法学习单个链接的条件概率而且使用自然的bayes假设。发明人发现,采用独立调控模型活动预测产生的群集最能被它们的存活分离(自然对数等级p=2.0x10-4)。发明人还利用具有针对p(xi|y)参数的平坦初始设置的独立调控模型执行该测试,而且发现它比之前开发的算法执行地更糟糕。再次,这表明学习方法要求在使用关于平台初始互动设置时丢失的互动的类型的先验知识。
图7通过以其关联分数对组织中的每个互动着色而且与其重要性成比例地设置其饱和度,示出了最显著的组织差异链接用途。最强差别g-分数被看出是针对关键癌症基因和复合体调控的链接,包括tp53、myc/max、hif1a/arnt、tap73a、e2f1和ppara-rxra。特别关注的是初始地在gbm[脑和kirc(肾)]和ov(卵巢)中的tap73a调控链接内就不同的ppara-rxra调控的链接,而且特别关注ucec(子宫内膜)中的更少程度。图8a和8b示出了用于激活来自ppara-rxra和tap73a的链接的组织所组的wpmi信号的曲线,其中在激活对角发现显著增大的重量,表明更多使用这些链接作为这些组织中的催化剂。如可从示出了针对具有ppara的链接的wpmi值的图8a看出:rxra作为父节点,gbm和kirc中存在更强激活信号,而且图8b示出了针对采用tap73a作为父节点的链接的wpmi值,表明ov中的激活。
tap73活动的签名潜在地表明与p73表达相关的通路起源的女性生殖或荷尔蒙模式。tap73促进了细胞循环抑制剂和细胞死亡诱导物的表达,其中一个是肿瘤抑制基因bax,其作为致癌基因bcl2的活动的抑制剂。已知在浆液卵巢癌症中高度表现bcl2,而且结果在此显示出,虽然tap73高度表现而且是bax表达的强促进剂(因此bcl2抑制),但是对于延迟肿瘤发生是无效的,暗示bcl2的小分子抑制可同等地无效。不令人惊讶的是,具有小分子抑制剂bcl2的卵巢癌症的单药治疗,哪怕高bcl2在浆液卵巢癌症中表达,还没有成功,暗示该类型癌症中的tap73-调停的活动的下游封锁或衰退。重要的是要注意,几乎所有浆液卵巢样本在此挖出p53中的突变,可能暗示肿瘤发生的上游分流,以及可能的是,结果tap73过度表现或增加活动。其它组还示出gbm和kirc中的ppara-rxra活动的重要性以及它们对非诺贝特(一种ppara兴奋剂)的敏感度。通过该分析识别的组织-特定信号看起来重申了进来的在当前tcga数据集合的环境下检测时显得独特的生物发现。
在整个tcga群上学习到的最明显链接(参见表格1)是大量已知癌症基因,包括叉头框转录因子a1、p53和雌性激素受体α。为了利用david对具有最高g-分数的50个互动中设计的基因执行基因集合富化,发明人利用它们的组分基因替换了系列和复合物。这产生了david从顶部的50个链接识别出的112个独特基因。对于大量相关kegg项,包括‘癌症中的路径’、‘细胞死亡’、‘jak-stat信令路径’和‘mapksignaling路径’以及大量不同癌症类型特定项,这些基因被发现被显著富化(p<1e-7)。发明人随后将该结果与在路径中链接的基因的基因表达关联处查看可以发现的情况进行比较。发明人需要通过pearson关联从平整路径取出顶部的200个基因表达对以得到可与本发明算法产生的集合相比拟尺寸的独特基因(n=119)。虽然两个基因集合针对基因本体术语为生物处理(goterm_bp_fat)产生了类似的富化,但是使用基因表达关联比使用学习的链接,使用远远更少的kegg项(20比46,fdr<0.05)和fdr。两个集合之间重叠的kegg项具有确定集合中的更低的fdr。为了确保系列和复合体在路径中的平坦,不偏置这些结果,发明人针对路径中的仅仅非系列、非复合体链接重复该分析,发现了类似结果(学习的链接发现20个kegg项,表达关联则是3个,fdr<0.05)。
表格1:整个tcga上的同生群具有最高g测试分数的常规链接
针对小雨le-323的所有链接的p-值
中间节点
发明人还比较乳房癌症的亚类型之间的链接的强度,以得到亚类型之间的调控差异的一些观点(参见表格2)。组织之间的该比较以及其它比较从来没有找到完全从激活至抑制切换方向的链接。相反,发明人经常观察到,链接打开或关闭(例如从强催化剂转化为中性)。由于方向很少改变,发明人发现简单地观看链接的g-分数重要性之间的差异是有益的。发明人使用g-分数的等级差来在组间进行比较以调节g-分数对样本尺寸的依赖性。具有最高等级差的许多链接具有相同的父节点。出于该理由,表格2示出了每个父节点的基础上的具有最高等级差的链接。在基础肿瘤中的顶部10个链接中的更强的9个中,hif1a是父节点,而且细胞腔a肿瘤中更强的顶部四个链接将cebpb作为父节点。
表格2:基础(n=92)或细胞腔a(n=218)乳房肿瘤中的p<0.05的常规链接,每个父节点的g-分数中的最高等级差异
注意:该表格中的所有等级差异的条件的p<4.8e-4。所有边缘边缘被注释为转录催化剂。整个表格是补充材料。
为了识别临床上相关的活动和链接强度,发明人检查雌性激素受体-正(er+)乳房癌症病人而且对链接g-分数和ipl执行tcga存活数据的正规的cox回归以识别出最佳数量的特征来最好地划分群。在最小的λ处,coxnet模型包含最好地划分er+乳房癌症病人(参见表格3)的九个特征。九个特征中的四个是链接g-分数,其示出了这些分数的独立效用作为潜在的预后指标。
表格3:与er+乳房癌症病人的存活相关的路径特征(边缘和节点)
注意:边缘通过→识别,而且找到的所有边缘被注释为路径中的转录催化剂。
cebpb和hif1a/arnt都出现在表格2和3中。cebpb是已经与肿瘤进程、不良预后和er负状态相关联的转录因子。而且,在表达式hsp90b1上,cebpb调控的并且在表格2中的热休克蛋白质已经与远处转移相关联而且降低具有良好预测的乳房癌症病人的总存活数。hsp90b1已经经过临床试验作为黑素瘤的免疫疗法。hif1a/arnt超表达在临床上与er-和pr-乳房癌症有关,其中剪接变体已经与减少的无远处转移生存相关。由于基础肿瘤一般是er-,而且细胞腔a肿瘤一般是er+,不同链接强度可由于基础肿瘤中的剪接变体的增大的发生率。基础和细胞腔之间的g-分数分级差异的顶部的两个链接是激活hk1和hk2(己糖激酶)的hif1a/arnt,hk2包括在葡萄糖代谢和细胞死亡中而且已经与来自乳房癌症的脑转移和较差的生存后颅骨切开术相关。这些发现表明通过在肿瘤亚类型之间进行对照以及通过搜索临床变量可预测的亚型内的链接发现有关系的链接的可行性。
由上可知,应该理解的是,设想的系统和方法允许多个组学数据组合以学习从文献组编的调控互动的强度和符号。调节独立的假设实现了模型复杂度的下降而且实现了采用现有数据集合对调控参数的有效估计。而且,发明人还表明,独立假设对于极大部分的细胞调控程序是有效的。此外,在独立假设不成立的情况下,设想的是,独立因子可由对共存调控程序更适当地建模的更复杂的因子代替。当这些学习的参数可应用时,通过简单地查看样本的群中的最强链接或者通过查看关注的表型之间的互动如何改变,可获得生物学观点。
还应该理解的是,虽然癌症亚类型使用不同互动,但是互动通常具有一致的符号,而无论何时其被用于特定肿瘤中。而且,学习的互动符号与数据库中的互动符号的一致,无论路径数据库中的biopax语言中注释的互动符号的各种方式如何,表示路径数据库已经成功地并真实地将文献中的成千上万的湿实验室实验编入目录。
此外,应该理解的是,共存调节器的独立性为模型推导和参数学习提供了计算上的优势,而且还有助于模型解释。调控模型的因子可分解性对应于自然对数的线性。然而,模型中的大量调节器是复合体,而且复形成因子是非线性噪声-max函数。因此,调节的非线性仍可在因子图中通过表示物理复配来进行编码。这将真实性引导为路径中的最平常的链接的物理解释:独立调节器的竞争性结合应该线性地组合,只要真实独立的物理实体已经被捕获作为复合体。如果该物理解释是真实的,那么测得的物理结合常数和确定的互动分数的相对强度之间应该存在对应关系。在独立假设不成立的情况下,很可能的是,存在潜在的共存因子,其可通过利用诸如p(y|x1,x2)之类的的因子来代替p(y|x1)p(y|x2)来建模。
由于设想的方法和系统能够区分组织亚类型之间的互动关联,发明人还设想一种将表示组织(例如,从肿瘤活体组织切片获得)的组学数据集合归类为属于亚型特定组织(例如,属于针对特定药物的抗治疗肿瘤)的方法。类似于前面讨论的方法,设想的方法首先经由组学输入接口获取表示组织的组学数据集合,随后针对组学数据集合导出路径模型中的调控节点的多个调控参数中的一组互动关联。由此导出的一组互动关联随后与和已知亚型特定组织相关的先验已知的一组互动关联进行匹配,而且在需要时,匹配随后被用于组学数据集合的归类(例如,代表已知亚型特定组织,这样将组织归类为属于亚类型)。因此,应该理解的是,设想的系统和方法将能够仅仅根据一个或多个互动关联签名来以亚型表征组织。在其它设想的组织亚类型中,尤其有利的亚类型包括抗药性组织、分生组织、药物治疗组织或组织的克隆变异体。
而且,由于设想的系统和方法实现了通过信令路径和/或路径网络的信号流的识别,所以可以理解的是,设想的系统和方法对于识别路径模型中的药物靶点也是有用的。这种识别通常包括步骤(a)经由组学输入接口获取表示组织的组学数据集合,(b)针对组学数据集合导出路径模型中的调控节点的多个调控参数中的一组互动关联,以及(c)在预测药物干扰了互动关联处将药物识别为影响了通路的活动。更典型地,调控节点影响了蛋白质的转录、转译和后转译改型中的至少一个,而且药物是商用药物而且具有已知模式的作用。
因此,由于路径的调控参数中的特定互动关联是已知的,路径模型中的目标路径现在可利用表示组织的组学数据集合而被容易地识别出来,以及针对组学数据集合导出路径模型中的调控节点的多个调控参数中的一组互动关联。在药物具有对互动关联的已知的效果时,药物则可被用来对准目标路径。例如,药物的已知效果可以是对致活酶的抑制效果、对受体的抑制效果、以及对转录的抑制效果中的至少一个。因此,在其它适当的目标路径之中,特别设想的目标路径包括钙/钙调蛋白调控路径、细胞激素路径、趋化因子路径、增长因子调控路径、荷尔蒙调控路径、map致活酶调控路径、磷酸酶调控路径和ras调控路径。根据路径分析的结果,治疗建议可基于识别出的路径。
而且,应该理解的是,无需在病人身上实际执行治疗,但是一旦路径的调控参数中的一个或多个特定互动关联已知,可仿真治疗。该仿真可被用来将治疗结果或识别多种药物预测至通过路径的相当低的信号。因此,设想的方法还包括一种经由电脑模拟仿真药物的治疗效果的方法,包括:获取具有多个路径元素的路径模型的步骤,其中至少两个元素经由具有调控节点的通路彼此耦接,所述调控节点根据多个调控参数控制沿通路的活动。设想的方法还包括识别出对至少一个调控参数的影响已知的药物的步骤;通过组学处理模块并基于药物的已知效果,经由电脑模拟而改变路径模型中的调控节点、活动和至少调控参数中的至少一个的另一步骤;以及确定路径模型中的改变的次生效应的又一步骤。更典型地,次生效应在路径模型中的另一调控节点、另一活动和另一调控参数中。更典型地,次生效应在路径模型中的另一调控节点、另一活动和另一调控参数中。
对于本领域技术人员而言明显的是,前面已经描述的修改之外的许多其它修改是可行的,而不脱离此处的发明构思。因此,并不限制本发明的主题,除非在所附权利要求的精神中予以限定。而且,在理解说明书和权利要求时,所有术语应该以与本文一致的最广泛的可能方式进行理解。具体地,术语“包括”和“包含”应该被解释为以非排除的方式指出元素、组件或步骤,表示所引述的元素、组件或步骤可出现或被使用或与没有明确指出的其它元素、组件或步骤组合。在说明书权利要求指出从由a、b、c…、和n组成的组中选自的物体中的至少一个,该说法应该被解释为要求来自该组的仅仅一个元素,而且不是a加n、或b加n等等。
- 还没有人留言评论。精彩留言会获得点赞!