一种基于多目标遗传算法的带假结核酸结构预测方法与流程
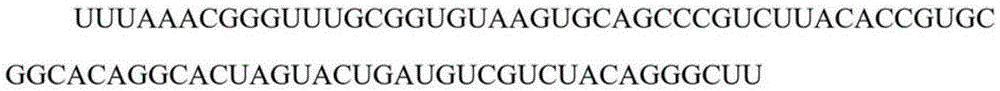
本发明属于生物信息工程领域,涉及一种核糖核酸(下文中简称rna)的二级结构预测的方法,尤其涉及基于多目标遗传算法的rna预测的方法。
背景技术:
rna序列的功能主要取决于它们的三维结构,但是通过rna分子一级结构直接预测其对应的空间结构十分困难。二级结构是由一级结构中碱基之间的配对和多核苷酸链自身折叠形成,其中不仅包括有序列信息,同时也含有三级空间结构信息。因此通过rna一级结构预测rna二级结构一直是研究rna整体结构的热点。
假结是rna序列中最广泛的结构单元,是非常复杂和稳定的rna结构。假结在rna序列中具有构造、催化和调节功能,是rna结构预测的关键点。目前试验方法是通过x射线衍射和核磁共振推断带假结的rna分子二级结构,这种方法虽然结果精确,但是只有在拥有相关设备的环境下才能进行,所用设备也非常昂贵且非常耗时。因此,采用计算机和热力学模型预测rna序列二级结构的方法被采用。
虽然不带假结的核酸分子二级结构预测已经有许多成熟的多项式算法,如动态规划算法;但是,对于含有假结的核酸分子的二级结构预测,迄今还没有有效的算法能够在多项式时间内求解,该问题已经被证实为np完全问题。因此现在急需一种可行的计算机高效方法来预测rna二级结构,降低时间和空间复杂度,特别是能够精确预测带假结的rna序列的二级结构。
带假结的rna二级结构预测如附图1、2所示,图1表示rna的一级结构序列,图2表示此rna对应的二级结构。
当前,国内外许多计算机科学家和生物学家提出了用于预测rna二级结构的方法,例如,pknotsrg-mfe方法、nupack方法,mfold方法等。其中mflod方法不能预测假结结构,且它的时间复杂度为o(n3)。rivas的pknotsrg-mfe方法处理了大量假结结构,提出一种基于最小自由能的动态规划算法模型,其时间复杂度为o(n4),空间复杂度o(n2),但是预测分子的最大长度不超过600。由dirks和pierce提出的nupack方法也是基于动态规划思想实现,该方法时间复杂度o(n5),空间复杂度o(n4)。由此可见,这些预测方法存在时间复杂度和空间复杂度偏高、仅能预测严格符合严格限制的rna二级结构且无法对长链的rna序列有效。
由于对rna二级结构预测方法的空间复杂度和时间复杂度直接影响预测成本,因此,如何使rna二级结构、尤其是包含假结的rna的二级结构的预测方法的时间复杂度和空间复杂度尽可能小,且确保预测结构的准确性,已经成为生物信息工程领域的一项重要的研究课题。
技术实现要素:
基于上述问题,本发明提出了如下的技术方案:
一种基于多目标遗传算法的带假结核酸结构预测方法,包括以下步骤:
s100,设置最小茎区数minstem、环中最小碱基数minloop、最大假结数maxpesudoknot,种群规模n,变异率pc,交叉率pm,最大进化代数gen进行初始化;
s101,长度为n的rna序列s表示为x1x2x3...xn,其中xi∈{a,c,g,u},1≤i≤n;对rna序列的每个碱基,用碱基所在位置序号来代替,表示为1,2,3,...,i,...,n的编码方式称为长度编码;
s102,判断待检测rna序列中的碱基配对情况,当rna序列中i,j位置发生碱基配对时,将对应的位置碱基编码进行交换;根据碱基配对情况计算符合碱基配对规则的随机点匹配列表:
(i,j,k),
其中i,j分别表示rna分子序列的第i个位置和第j个位置可以进行匹配,k为随机碱基对(i,j)的连续匹配数,得到随机点(i,j)的k连续匹配集(i,j,k1,k2,k3,...,kn)用以表示位置i和位置j匹配时可选的连续匹配数集合;
s103,使用模拟退火算法从连续匹配数集合中随机产生n个随机rna序列,设为初始种群p0;
s104,对当前种群pt进行遗传操作,包括对种群中n个rna序列进行选择、交叉和变异,得到子种群qt;
s105,将父代种群pt和子种群qt合并成为整体种群rt;
s106,对整体种群rt根据评价函数进行非支配排序构造出不同等级的非支配解集z1,z2,z3...,对分配好等级的非支配解集进行拥挤距离排序;
其中评价函数定义如下:f=(总的碱基匹配数,总分组数)
s107,根据排序的高低挑选出前n个解,构成下一次迭代的父代种群pt+1;
s108,判断进化代数是否达到设定的最大值gen,如果达到最大值则进入步骤s109,否则进入步骤s104;
s109,输出pareto最优解集。
s110,计算pareto最优解集中所有rna分子的自由能,输出自由能最小的rna分子结构。
优选地,步骤s102中随机点匹配列表(i,j,k)中随机数i,j需要同时满足以下关系:
i<j
j-i-k>3
最小茎区数≤k≤2/3*序列长度n
若随机数不满足上述关系,则重新生成随机数,若满足,则判断是否满足k连续匹配,若不满足k连续匹配,则重新生成随机点,满足,则添加到随机点匹配列表中。
优选地,步骤s102生成随机点匹配列表(i,j,k)后对其进行k连续匹配验证,步骤如下:
首先对单独位置上的碱基组合依照watson-crick碱基配对规则进行校验,先把分子序列重新编码,编码规则按照a,c,g,u依次对应0,1,2,3,根据碱基配对规则,若第i位置与第j位置为基本匹配,即a-u,g-c或u-a,c-g配对时,需要满足如下条件:
rnaseq[i]+rnaseq[j]=3
当匹配为g-u或u-g配对时,则需满足以下条件:
rnaseq[i]+rnaseq[j]=5
当从第i位置开始到第i+k-1位置为止分别与第j位置开始到第j-k+1位置为止,均满足上述条件,则随机生成的三元组(i,j,k)满足k连续匹配。
本发明的基于多目标遗传算法的带假结核酸结构预测方法,通过最小茎区数和环中最小碱基数来确定随机点k连续匹配列表,生成茎区候选区域;再利用了多目标遗传算法的思想,使得更快的产生有效匹配;最后引入能量评价函数,来提高rna分子假结预测的准确率,从而降低了时间复杂度和空间复杂度,以及提高了rna分子假结预测的准确率。本发明的预测方法,可有效降低预测成本,使之广泛应用于生物信息工程领域。
附图说明
图1为rna的一级结构序列实例。
图2为图1中rna一级结构序列对应的二级结构。
图3为本发明的带假结核酸结构预测方法实施例的流程示意图。
图4为本发明实施例中rna序列长度编码示意图。
图5为图4中的rna序列长度编码对应的rna序列实例。
图6为本发明实施例中交换个体parentx和parenty在i和j之间的对应片段,生成新的个体offspringx,offspringy的示意图。
图7为本发明实施例中在连续匹配集中重新选择i和j之间的对应片段的连续匹配数k,生成新的个体offspring的示意图。
图8示出本发明的方法与采用pknotsre算法的技术方案的sensitivity和specificity对照表。
具体实施方式
为使本发明更加容易理解,下面结合附图和实施例进一步说明本发明的技术方案。
如图3所示,本发明的基于多目标遗传算法的带假结核酸结构预测方法的一种实施例,包括以下步骤:
s100,设置最小茎区数minstem、环中最小碱基数minloop、最大假结数maxpesudoknot,种群规模n,变异率pc,交叉率pm,最大进化代数gen进行初始化。
作为优选实施方案,本实施例中,最小茎区数设置为2,由于rna序列不能剧烈折叠,环中都需要至少间隔三个碱基,因此环中最小碱基数需默认设置为3,当rna分子中碱基数量为500以下时,最大假结数设置为1,碱基数量为500-1000时,最大假结数设置为2或3,种群规模n设置为100,变异率设置为0.01,交叉率设置为0.8,最大进化代数gen设置为1000。
s101,长度为n的rna序列s表示为x1x2x3...xn,其中xi∈{a,c,g,u},1≤i≤n;对rna序列的每个碱基,用碱基所在位置序号来代替,表示为1,2,3,...,i,...,n的编码方式称为长度编码;
s102,判断待检测rna序列中的碱基配对情况。对单独位置上的碱基组合依照watson-crick碱基配对规则进行校验,先把分子序列重新编码,编码规则按照a,c,g,u依次对应0,1,2,3,根据碱基配对规则,若第i位置与第j位置为基本匹配,即a-u,g-c或u-a,c-g配对时,需要满足如下条件:
rnaseq[i]+rnaseq[j]=3
当匹配为g-u或u-g配对时,则需满足以下条件:
rnaseq[i]+rnaseq[j]=5
当从第i位置开始到第i+k-1位置为止,分别与第j位置开始到第j-k+1位置为止,均满足上述条件,则随机生成的三元组(i,j,k)满足k连续匹配。即rna序列长度可编码为:1,2,3,...,j,j-1,...j-k+1,...,i+k-1,...,i+1,i,...,n,如图4,5所示。
此外,随机点匹配列表(i,j,k)中随机数i,j需要同时满足以下关系:
i<j
j-i-2*k>3
最小茎区数≤k≤2/3*序列长度n
若随机数不满足上述关系,则重新生成随机数,若满足,则判断是否满足k连续匹配,若不满足k连续匹配,则重新生成随机点,满足,则添加到随机点匹配列表中。
当rna序列中i,j位置发生碱基配对时,将对应的位置碱基编码进行交换;根据碱基配对情况计算符合碱基配对规则的随机点匹配列表:
(i,j,k),
其中i,j分别表示rna分子序列的第i个位置和第j个位置可以进行匹配,k为随机碱基对(i,j)满足的连续匹配数,得到随机点(i,j)的k连续匹配集(i,j,k1,k2,k3,...,kn)用以表示位置i和位置j匹配时可选的连续匹配数集合。
s103,使用模拟退火算法从连续匹配数集合中随机产生n个随机rna序列,设为初始种群p0;
s104,对当前种群pt进行遗传操作,包括对种群中n个rna序列进行选择、交叉和变异,得到子种群qt;
选择:即从当前种群pt中选择排序靠前的n个个体
交叉:随机产生两个实数i,j,其中1≤i≤j≤n,然后随机产生一个概率p,随机选取种群pt中的两个个体parentx和parenty。如果p>pc,则交换个体parentx和parenty在i和j之间的对应片段,生成新的个体offspringx,offspringy,如图6所示,否则不发生交叉。
变异:随机产生两个实数i,j,其中1≤i≤j≤n,然后随机产生一个概率p,随机选取种群pt中的一个个体parent。如果p>pc,则从连续匹配集中重新选择i和j之间的对应片段的连续匹配数k,生成新的个体offspring,如图7所示,否则不发生变异。
s105,将父代种群pt和子种群qt合并成为整体种群rt;
s106,对整体种群rt根据评价函数进行非支配排序构造出不同等级的非支配解集z1,z2,z3...,对分配好等级的非支配解集进行拥挤距离排序;
其中评价函数定义如下:f=(总的碱基匹配数,总分组数)
定义个体i总的碱基匹配数多于个体j总的碱基匹配数并且i的总分组数小于j的总分组数则称fi>fj,即个体i支配个体j。用ni表示种群中所有个体中支配个体i的数目,si表示种群中个体被个体i支配的个体集合,则非支配排序过程可表示为:
1、找出种群中非支配解的个体,即ni=0的个体,将非支配个体放入集合f1中。
2、对于f1中的每个个体,找出集合中每个个体所支配个体集合si,对si中的个体1,对n1进行减1操作,令n1=n1-1,若n1大小为0,则将此个体存放在集合h中。
3、定义集合f1为第一层非支配集合,并为f1中每个个体标记相同的非支配序列irank。
4、对集合h中的个体,按照以上步骤1、步骤2和步骤3操作,直至将所有个体分层。
对种群中所有个体进行非支配排序后,对同一层级通过拥挤距离进行排序,设f1总的碱基匹配数,f2为总分组数,则同一非支配层级中个体i的拥挤距离可表示为:
l[i]=|f1(i+1)-f1(i-1)|+|f2(i+1)-f2(i-1)|
当每个个体拥有这两个属性,就可以通过这两个属性判定任意两个个体的支配关系。当两个个体没有处于同一非支配层级时,通过判断irank大小,确定个体优劣,irank值小的个体比irank大的个体更优;当两个随机个体处于同一个非支配层级时,依据个体拥挤距离判断个体优劣,其中拥挤距离大的个体比拥挤距离小的个体更优。
s107,根据排序的高低挑选出前n个解,构成下一次迭代的父代种群pt+1;
s108,判断进化代数是否达到设定的最大值gen,如果达到最大值则进入步骤s109,否则进入步骤s104;
s109,输出pareto最优解集;
s110,计算pareto最优解集中所有rna分子的自由能,输出自由能最小的rna分子结构,即当前分子的随机点匹配列表(茎区列表)。
如上所述,即使rna序列中包含了非嵌套结构和假结结构,本发明的方法依旧能够较准确地确定rna的二级结构。
图8示出本发明与采用pknotsre算法的技术方案的sensitivity和specificity对照表。在图8中,sensitivity=tp/rp,specificity=tp/(tp+fp),其中tp表示rna结构中正确预测的碱基对数量,fp表示rna结构中错误预测的碱基对数量,rp表示rna结构中真实的碱基对的数量。可以看出,本发明的预测方法具有较好的预测准确度,且具有较低的时间复杂度和空间复杂度。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!