多元互信息和残基结合能量蛋白质间相互作用预测方法与流程
本发明涉及生物信息技术中基于氨基酸序列信息对蛋白质与蛋白质之间的相互作用进行预测的方法,属于蛋白质组学中的大分子结构预测算法领域。具体讲,涉及多元互信息和残基结合能量蛋白质间相互作用预测方法。
背景技术:
蛋白质与蛋白质之间的相互作用是许多生物过程的核心。识别蛋白质之间的相互作用对于阐明蛋白质功能和鉴定细胞中的生物过程是非常重要的。蛋白质间的相互作用信息可以帮助人们更好地了解疾病发生机制,从而更加高效准确的进行药物设计。在过去的几年中,大量的计算技术已经发展到可以进行大规模分析的阶段。一般来说,检测蛋白质间相互作用的计算方法主要有三类:基于进化信息的方法,基于自然语言处理的方法和基于氨基酸序列特征的方法。基于进化信息的方法从同源蛋白的多重序列比对中提取进化信息,构建进化树来分析蛋白质功能之间的关系。该方法需要大量同源蛋白质数据和这些蛋白质之间的相互作用标记,因此其在大规模的计算使用上受到了很大限制。基于自然语言处理的方法依托于具有广泛应用的自然语言处理技术。此类方法从存储在生物学和医学科学文献中的大量已知的蛋白质间相互作用关系中挖掘有用信息。由于文献中部分信息的缺少,预测结果可能并不完整。因此,采用基于氨基酸序列的多元互信息特征提取方法和残基结合能量信息特征提取方法来提高蛋白质之间相互作用的预测精度和保证方法的大规模推广使用就显得尤为重要。
作为基于氨基酸序列信息进行蛋白质之间相互作用预测方法的关键技术,特征提取方法是指定义一系列的映射函数,通过这些函数将蛋白质的一段氨基酸序列中映射成一列能够代表该序列的特征数值。这些数值要尽可能全面的包含蛋白质的有用的特征,同时要排除掉会对预测结果产生不利影响的噪音信息。经典的氨基酸序列特征提取方法包括自协方差、联合三联体、局部蛋白质序列描述子、多尺度局部特征描述符、局部相位量化描述符以及基于矩阵的蛋白质序列表示等方法。这些方法从不同方面对氨基酸序列进行了抽象表示,同时其预测结果有着很大的差异。因此如何设计一种有效的特征提取方法来抽象映射氨基酸序列,提高序列之间的可区别度,降低噪音信息对预测结果的干扰成为蛋白质间相互作用预测方法的关键技术。
技术实现要素:
为克服现有技术的不足,本发明旨在提出一种能够准确高效的预测蛋白质与蛋白质之间相互作用的方法。该方法所使用的特征提取函数能够提高氨基酸序列中有用信息在预测操作中的作用,同时有效降低无用噪音信息的影响。为此,本发明采取的技术方案是,多元互信息和残基结合能量蛋白质间相互作用预测方法,步骤如下:
步骤(1):氨基酸类别分组,将20种标准氨基酸按照偶极性和体积分到n个功能组,这n个功能组分别记为c0,c1,c2,…,cn,将原始氨基酸序列按照各个氨基酸所在的功能组类别转换成组类别序列;
步骤(2):定义不同类型的3元组和2元组特征表示,3元组的特征表示为”c0c0c0”,“c0c0c1”,…“cncncn”;2元组的特征表示为”c0c0”,”c0c1”,…”cncn”。
步骤(3):统计组类别序列中,3元组特征和2元组特征出现的个数,建立特征频数表,使用频率计算函数f(a)=(na+1)/(l+1)分别计算n个类别在序列中出现的频率;
步骤(4):计算2元组互信息特征,计算公式为:
其中f(ab)是二元组中同时出现类别ab的频率;
步骤(5):计算3元组互信息特征。计算公式为:
i(abc)=i(ab)+f(a|c)lnf(a|c)
-f(a|bc)lnf(a|bc)
其中f(a|c)是所有出现类别c的二元组中同时出现类别a的频率,f(a|bc)是所有出现类别bc的三元组中同时出现类别a的频率;
通过上述5个步骤得到第一部分互信息特征值;
步骤(6):计算氨基酸理化属性特征;
步骤(7):通过统计分析蛋白质复合物数据库,使用残基配对频率计算氨基酸接触矩阵aac:
其中i,j表示两种氨基酸,ni,j=∑dnij是i和j的接触数量,
计算替代矩阵smr,smri,l=aac(i,al),其中i=1,…,20是二十种氨基酸类型之一,l=1,…,l是给定蛋白质序列中l个位置之一,al是l位的氨基酸类型,通过该步骤得到一个20×l的替代矩阵smr;
步骤(8):使用梯度方向直方图hog特征提取算法对氨基酸序列进行特征提取;
步骤(9):对smr矩阵的转置矩阵进行奇异值分解,通过奇异值分解可以得到20个右奇异向量。
步骤(10):将通过步骤1至9得到的特征值输入到一个随机森林模型进行预测,从而得到两条蛋白质之间的相互作用。
步骤(6)具体计算步骤如下:
步骤(6.1):计算moreau-broto自相关特征值,计算公式为:
其中lag是残基之间的距离,p是上述自然氨基酸的第p个物理化学性质,l是序列的位置,l=1,2,...,l-lag,且lag=1,2,...,lg,用六种理化性质表示之后,得到lg×6个特征值。
步骤(6.2):将得到的lg×6个特征值进行归一化处理;
步骤(6.3):统计序列上20个氨基酸出现的频率。
步骤(8)具体计算过程如下:
步骤(8.1):计算水平和垂直方向的梯度值gh(i,l)、gv(i,l),计算公式为:
步骤(8.2):计算梯度幅值
步骤(8.3):计算梯度方向
步骤(8.4):将梯度幅值矩阵和梯度方向矩阵分割为9个相同大小的子矩阵;
步骤(8.5):统计各个梯度方向的直方图,每一个梯度方向的直方图大小作为一个特征值。
通过上述步骤,每条序列得到x个特征值,两条序列一共得到2x个特征值。
本发明的特点及有益效果是:
由于本发明集成了氨基酸序列的多元互信息和残基结合能量信息。与传统的序列信息相比,多元互信息既考虑了每个氨基酸伴随其两个邻位肽氨基酸的特性,又考虑了其组分的互信息。同时梯度直方图和奇异值分解操作能够提取蛋白质矩阵的纹理特征。这些新的信息和特征的加入,为准确预测蛋白质间的相互作用关系提供了有力的帮助,因而本方法在对蛋白质与蛋白质相互作用关系进行分析和预测时,预测结果的准确性比现有的其他方法更优。本方法不仅能准确预测蛋白质间的相互作用,同时还能在蛋白质相互作用网中发现新的相互作用关系,对完善各类蛋白质相互作用网络有着很大的意义。
附图说明:
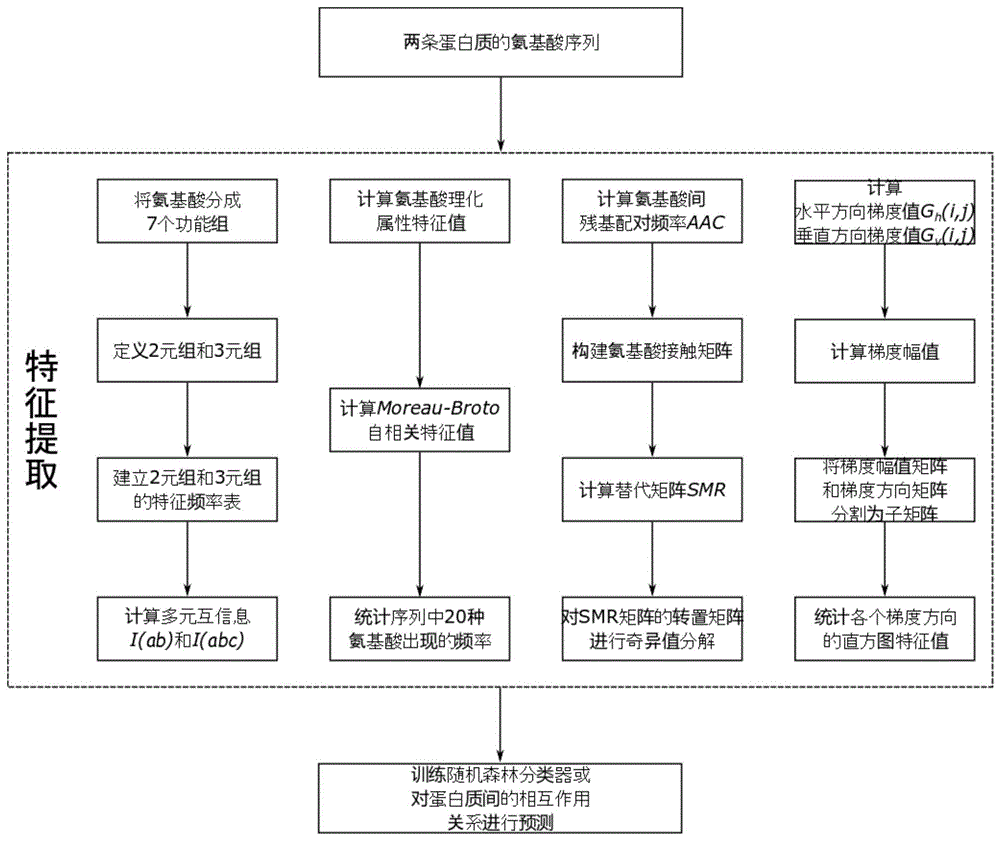
图1.本发明的计算过程的流程图
图2.二元组和三元组的特征表示及频率表的建立;
图3.计算梯度方向直方图的示意图;
图4.moreau-broto自相关特征在使用不同lg值时的准确率;
图5.本方法在单核心网络上的预测结果;
图6.本方法在多核心网络上的预测结果;
图7.本方法在交叉网络上的预测结果。
具体实施方式
本发明的目的在于提供了一种能够准确高效的预测蛋白质与蛋白质之间相互作用的方法。该方法所使用的特征提取函数能够提高氨基酸序列中有用信息在预测操作中的作用,同时有效降低无用噪音信息的影响。
本发明的特点在于,它依次含有以下步骤:
步骤(1):氨基酸类别分组。将20种标准氨基酸按照偶极性和体积分到7个功能组。这7个功能组分别记为c0,c1,c2,...,c6。将原始氨基酸序列按照各个氨基酸所在的功能组类别转换成组类别序列。
步骤(2):定义不同类型的3元组和2元组特征表示。3元组的特征表示为”c0c0c0”,“c0c0c1”,…“c6c6c6”。2元组的特征表示为”c0c0”,”c0c1”,…”c6c6”。
步骤(3):统计组类别序列中,3元组特征和2元组特征出现的个数,建立特征频数表,如图2所示。使用频率计算函数f(a)=(na+1)/(l+1)分别计算7个类别在序列中出现的频率。
步骤(4):计算28个2元组互信息特征。计算公式为:
其中f(ab)是二元组ab出现的频率。
步骤(5):计算84个3元组互信息特征。计算公式为:
i(abc)=i(ab)+f(a|c)lnf(a|c)
-f(a|bc)lnf(a|bc)
其中f(a|c)是所有出现类别c的二元组中同时出现类别a的频率,f(a|bc)是所有出现类别bc的三元组中同时出现类别a的频率。
通过上述5个步骤可以得到238个互信息特征值。
步骤(6):计算氨基酸理化属性特征。每条氨基酸序列可以得到200个特征值,一对要预测相互作用的氨基酸序列可以得到400个特征值。具体计算方法如下:
步骤(6.1):计算moreau-broto自相关特征值。计算公式为:
其中lag是残基之间的距离,p是上述自然氨基酸的第p个物理化学性质,l是序列的位置,l=1,2,...,l-lag,且lag=1,2,...,lg,这里的lg一般取值为30。用六种理化性质表示之后,可以得到30×6=180个特征值。
步骤(6.2):将得到的180个特征值进行归一化处理。
步骤(6.3):统计序列上20个氨基酸出现的频率。
步骤(7):通过统计分析蛋白质复合物数据库,使用残基配对频率计算氨基酸接触矩阵aac:
其中i,j表示两种氨基酸。ni,j=∑dnij是i和j的接触数量。
计算替代矩阵smr,smri,l=aac(i,al),其中i=1,…,20是二十种氨基酸类型之一,l=1,…,l是给定蛋白质序列中l个位置之一,al是l位的氨基酸类型,通过该步骤得到一个20×l的替代矩阵smr;
步骤(8):使用梯度方向直方图hog特征提取算法对氨基酸序列进行特征提取,具体计算过程如下:
步骤(8.1):计算水平和垂直方向的梯度值gh(i,l)、gv(i,l),计算公式为:
步骤(8.2):计算梯度幅值
步骤(8.3):计算梯度方向
步骤(8.4):将梯度幅值矩阵和梯度方向矩阵分割为9个相同大小的子矩阵。
步骤(8.5):统计各个梯度方向的直方图。每一个梯度方向的直方图大小作为一个特征值。
通过上述步骤,每条序列可以得到81个特征值,两条序列一共可以得到162个特征值。
步骤(9):对smr矩阵的转置矩阵进行奇异值分解。通过奇异值分解可以得到20个右奇异向量。该步骤可以得到800个特征值。
步骤(10):通过步骤1至9,一共可以得到238+400+162+800=1600个特征值。将这些特征值输入到一个随机森林模型进行预测,从而得到两条蛋白质之间的相互作用。
按照上述计算方法,我们使用前人构建的12个研究人员普遍认可的蛋白质与蛋白质相互作用数据集通过随机森林模型对我们的预测方法进行了性能上的分析。其中包括s.cerevisiae、h.pylori2918、human8161和e.coli等数据集。同时还将该方法在三个真实的蛋白质相互作用网络上进行了试验分析,例如单核心网络cd9、多核心网络ras-raf-mek-erk-elk-srf代谢路径以及交叉网络wnt。在s.cerevisiae数据集上,使用二元互信息、三元互信息和多元互信息来进行相互作用预测所达到的准确率分别为93.56%、93.88%和94.23%。显而易见,使用组合的多元互信息进行特征提取比单独使用一类特征提取能获得更好的性能。对于moreau-broto自相关特征值计算方法,我们为了获得最好的lg,本文测试了九个不同的lg值(lg=5,10,15,20,25,30,35,40,45)。图4显示了当分别使用不同的lg值时预测结果的准确性。从图中曲线可以看出,当lg从5增加到30时,预测精度增加,然而,当lg从30增加到45时,精度在降低。最佳预测精度在lg为30时获得,准确率为92.76%。方法中使用的梯度方向直方图和奇异值分解两类特征值单独使用时所能达到的准确率分别为93.86%和92.93%。在我们的方法中,四类特征提取方法集成起来,预测的准确率为94.56%。方法中使用的随机森林分类器要比支持向量机分类器在预测结果上有优势。随机森林分类器为集成模型,同时能够检测特征的重要性,因此在预测结果的准确度上有2%的提高。
该方法应用于蛋白质相互作用网络预测时,也有很高的准确率。在单核心网络cd9上,我们的方法可以识别16个蛋白质相互作用关系中的14个,准确率为87.50%。ras-raf-mek-erk-elk-srf代谢路径的多核心网络上,我们可以正确的预测出189个蛋白质相互作用关系中的174个,准确率为92.06%。与wnt相关代谢路径的交叉网络在信号传导中至关重要。我们的方法从96个相互作用关系中发现了91个,准确率为94.79%,优于已有的预测方法。当前已有的其他方法在这三类网络结构上,所能达到的准确度分别为81.25%、90.00%和76.04%。由此可见,我们的方法比已有的方法有更高的准确度。图5、6、7是我们的方法在但核心网络、多核心网络和交叉网络上进行蛋白质间相互作用关系的发现和预测时所得到的结果。图中蓝线表示正确的预测,红线表示错误的预测。从这3个图中可以看出,我们的方法具有很高的可信性和实用性。
在蛋白质组学中,对蛋白质间相互作用预测问题的最大困难是已有信息不够清晰,有用的信息都隐含在过于简单的序列信息中。如果要准确的对相互作用进行预测,不能仅仅使用直接的序列信息,而是需要有好的信息提取方法从底层序列信息中抽象出更加丰富有用的交互信息和理化属性信息。对于蛋白质间相互作用的预测问题,设计一种通用的能够从序列信息中提取出有用信息的特征提取方法,是本发明的主要贡献。
该发明的基本思想是:提取融合多种类型的属性信息,通过有效的分类器对相互作用进行预测。该发明首先计算各类氨基酸以及其组成的二元组和三元组在序列中出现的频率信息,然后在这些频率信息的基础上,进一步整合抽象出多元互信息,从简单的序列数据中挖掘不同氨基酸及其元组之间存在的联系。其次,该发明还充分考虑氨基酸的理化性质对相互作用的影响,从序列中提取出残基结合能量信息来进一步提高预测的准确度。
该发明主要包含以下步骤:计算氨基酸序列中的多元互信息,得到238个互信息特征值。计算moreau-broto自相关特征值并统计20中氨基酸在序列中出现的频率,得到400个特征值。使用残基配对频率计算氨基酸接触矩阵,然后计算替代矩阵。通过使用梯度方向直方图对替代矩阵进行处理,可以得到162个特征值。同时对替代矩阵进行奇异值分解可以得到800个特征值。将得到的1600个特征值使用随机森林分类器进行分类,从而判断两个蛋白质之间是否存在相互作用。
该发明的计算过程具有简单易于实现的特点,并且计算所需的硬件设备和计算资源也比较低,具有广泛的可使用性。我们的方法可以通过c++和matlab来实现,在普通的2.5ghz6核cpu和32gb内存的计算机上,对数千个样本进行预测的任务能够在很短的时间内完成。同时,为了平衡性能和效果之间的关系,随机森林分类器的决策树数量和每颗子树可利用特征数分别选取为500和400。通过调整这些参数,还可以提高分类计算的速度从而更快的进行预测操作。
- 还没有人留言评论。精彩留言会获得点赞!