含有膦酰基-磷酸根的化合物和聚合物的制作方法
本发明涉及含有膦酰基-磷酸根的新型化合物和聚合物。本发明还涉及使用新型化合物和聚合物来处理表面的方法。
背景技术:
与溶液中的多价阳离子以及与包含多价阳离子的表面相互作用的化学结构可用于这些体系的处理。例如,多磷酸盐和焦磷酸盐已被用作衣物洗涤和餐具制剂中的助洗剂以控制钙,并且用于钻探泥浆中以防止沉淀。它们也已被用于口腔护理行业中以帮助控制牙垢并降低牙齿上菌膜层的厚度,从而获得牙齿光滑感。类似地,由于双膦酸盐和羟基-双膦酸盐与钙羟基磷灰石表面的强相互作用,双膦酸盐和羟基-双膦酸盐是骨质疏松药物中的活性组分,并且还用作盘碟洗涤液和锅炉系统中的晶体生长抑制剂。这些示例中的每一个都有着固有的局限性。多磷酸盐易于在所有ph下随时间推移在水溶液中降解,最终导致溶液中的正磷酸根增加。多磷酸盐也是相当阴离子性质的,并且不溶于非极性有机体系。然而,多磷酸盐通常可安全食用,并且可用于不同的食物产品中。相反,双膦酸盐和羟基-双膦酸盐在水中长期稳定,并且可根据连接到双膦酸盐碳上的有机基团的性质,使其在有机体系中完全溶解。然而,双膦酸盐具有骨活性,由于其强效药理作用,因此不能用于食物或其他可能被意外摄入的系统中。包含分子量不足以穿过肠壁的双膦酸盐的聚合物将可能不具有骨活性,然而可穿过肠壁的任何低分子量残余单体或低聚物使得此类聚合物在潜在可摄入环境中禁用。此外,由于双膦酸盐不易分解,因此它们的活性在使用后可在环境中持续存在。
因此,仍然需要不易降解并且对于人类食用是安全的磷酸盐组合物。
技术实现要素:
已惊奇地发现,膦酰基-磷酸根化学基团改善了对多磷酸盐和双膦酸盐的关注,同时发现了在类似体系中的实用性。具体地讲,无论是通过掺入含膦酰基-磷酸根基团的单体还是通过后聚合改性来添加膦酰基-磷酸根基团,包含膦酰基-磷酸根基团的组合物、包含膦酰基-磷酸根基团的单体以及包含膦酰基-磷酸根基团的聚合物均可用于其中使用含有多磷酸盐和双膦酸盐的结构的许多应用中。此类应用通常包括其中结合相互作用涉及溶液中和含有二价阳离子的表面上的多价阳离子的那些。含膦酰基-磷酸根的结构也可用于多磷酸盐和双膦酸盐用途有限的应用中。膦酰基-磷酸根基团是条件稳定的,并且将仅在酸性或催化条件下释放磷酸盐。因此,膦酰基-磷酸根基团比多磷酸盐更稳定,但不如双膦酸盐稳定。这使得能够配制成这样的体系,其中食用无害性和水稳定性为必须要求。此外,连接到膦酰基-磷酸根基团的有机基团或聚合物可导致整个分子可溶于有机溶剂,或用于向整个分子添加额外的官能度。
在一个实施方案中,本发明涉及具有式1的结构的化合物:
其中:
r1选自-h和-ch3;
r2选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式2的结构:
其中:
δ为与式1的连接位点,
r5和r6独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;
r3选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式3的结构:
其中:
δ为与式1的连接位点,
r7和r8独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
n为1至22的整数;
r4选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
l选自化学键、芳二基和式4的结构:
其中:
α为与链烯基基团的连接位点;
β为与膦酰基-磷酸根基团的连接位点;
x选自式5至式11所示的结构;
其中:
r9选自-h、烷基(c1-8)、膦酰基烷基和膦酰基(磷酸)烷基;并且
y选自链烷二基、烷氧基二基、烷基氨基二基和链烯二基。
在化合物的一个实施方案中,式1的r1为h。在化合物的另一个实施方案中,式1的r1为ch3。在化合物的另一个实施方案中,式1的l为共价键。
在化合物的一个实施方案中,r2、r3和r4独立地选自-h、na盐和k盐。在化合物的另一个实施方案中,l具有式4的结构,并且x具有式5的结构。在化合物的另一个实施方案中,l具有式4的结构,并且x具有式8的结构。在化合物的一个实施方案中,l具有式4的结构,并且x具有式10的结构。
本发明的另一个实施方案为一种新型聚合物。该聚合物包含膦酰基-磷酸根基团,其中该膦酰基-磷酸根基团具有式12的结构:
其中:
ε为与所述聚合物的主链、侧基或侧链中的碳原子的连接位点;
r10选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式13的结构:
其中:
θ为与式12的连接位点,
r13和r14独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;
r11选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式14的结构:
其中:
θ为与式12的连接位点,
r15和r16独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
n为1至22的整数;并且
r12选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐。
在一个实施方案中,用于形成该聚合物的至少一种单体包含膦酰基-磷酸根基团。在另一个实施方案中,在后聚合改性过程中加入该膦酰基-磷酸根基团。
在另一个实施方案中,用于形成该聚合物的至少一种单体具有式15的结构:
其中:
ω为与式12的膦酰基-磷酸根基团的连接位点;
r17选自-h和-ch3;
l1选自化学键、芳二基和式4的结构:
其中:
α为与链烯基基团的连接位点;
β为与式12的膦酰基-磷酸根基团的连接位点;
x选自式5至式11中的结构;
其中:
r9选自-h、烷基(c1-8)、膦酰基烷基和膦酰基(磷酸)烷基;并且
y选自链烷二基、烷氧基二基、烷基氨基二基和链烯二基。
在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为h。在另一个实施方案中,当用于形成聚合物的至少一种单体具有式15的结构时,r17为ch3。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1为共价键。
在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r10、r11和r12独立地选自-h、na盐和k盐。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式5的结构。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式8的结构。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式10的结构。
通过阅读本公开内容,本发明的这些和其它特征、方面和优点将对于本领域的技术人员变得显而易见。
附图说明
图1为示出聚合物性能的图表。
图2为示出聚合物性能的图表。
图3为示出聚合物性能的图表。
图4为示出聚合物性能的图表。
图5为由聚合物分析得到的gpc迹线图。
图6为由聚合物分析得到的gpc迹线图。
具体实施方式
虽然说明书最后由权利要求书具体地指出并清楚地要求保护本发明,但据信本发明将由以下说明更好地理解本发明。
除非另外指明,本文所有百分数是按组合物的摩尔计。
除非另外指明,所有比率均是摩尔比。
除非另外指明,本文提及的成分的所有百分比、比率和含量均基于该成分按摩尔计的实际含量,并且不包括在市售产品中可与这些成分组合使用的溶剂、填料或其他材料。
如本文所用,“包括/包含”是指可加入不影响最终结果的其它步骤和其它成分。该术语涵盖术语“由……组成”和“基本上由……组成”。
所有引用的参考文献均全文引入本文以供参考。任何文献的引用并不是对其作为受权利要求书保护的本发明现有技术的可获得性的认可。
定义
术语“位点”或“连接位点”或“连接点”均是指在化学基团或限定的结构实体内具有开放价态的原子,该原子被指定带有符号诸如简单的短横线(-)或后接短横线或直线的小写希腊字母(例如,α-、β-等),以指示如此指定的原子经由化学键连接至独立化学基团中的另一原子。符号
还指示化学基团的连接点。需注意,这种方式通常仅用以识别较大化学基团的连接点,以便明确地帮助读者识别与化学键从其延伸的原子的连接点。第一化学基团或限定的结构实体上的连接位点或连接点通过单键、双共价键或三共价键连接至第二化学基团或限定的结构实体上的连接位点或连接点,以便满足所连接的原子的正常化合价。
当与化学基团一起使用时,术语“基团”表示任何连接的原子基团,诸如甲基基团、羧基基团或作为较大分子的一部分的膦酰基-磷酸根基团。
当在化学基团的语境中使用时:“氢”是指-h;“羟基”是指-oh;“氧代基”是指=o;“羰基”是指-c(=o)-;“羧基”和“羧化物”是指-c(=o)oh(也写为-cooh或-co2h)或其去质子化形式;“氨基”是指-nh2;“羟氨基”是指-nhoh;“硝基”是指-no2;“亚氨基”是指=nh;“氧化胺”是指n+o-,其中n与除了o之外的原子具有三个共价键;“异羟肟酸”或“异羟肟酸根”是指-c(o)nhoh或其去质子化形式;在一价的语境中,“磷酸根”是指-op(o)(oh)2或其去质子化形式;在二价的语境中,“磷酸根”是指-op(o)(oh)o-或其去质子化形式;“膦酸根”是指c-p(o)(oh)2或其去质子化形式,其中c具有正常的四价和三个与除了p之外的原子连接的共价键;“膦酰基-磷酸根基团”是指通过共享氧原子与至少一个磷酸根化学键合的膦酸根,诸如但不限于膦酰基-单磷酸c-p(o)(oh)op(o)(oh)2、膦酰基-二磷酸c-p(o)(op(o)(oh)2)op(o)(oh)2、膦酰基-环二磷酸
对于以下化学基团和类别,以下括号内的下标进一步将化学基团/类别定义如下:“(cn)”定义化学基团/类别中碳原子的确切数目(n)。“(c≤n)”定义了可在化学基团/类别中的碳原子的最大数目(n),其中所考虑的化学基团的最小数目尽可能小,例如,应当理解,化学基团“链烯基(c≤8)”或化学类别“链烯(c≤8)”中的碳原子的最小数目为2。例如,“烷氧基(c≤8)”表示具有1至8个碳原子的那些烷氧基基团。(cn-n’)定义了化学基团中碳原子的最小数目(n)和最大数目(n')两者。类似地,烷基(c2-8)表示具有2至8个(包括2个和8个)碳原子的那些烷基基团。
术语“阳离子”是指具有净正电荷的原子、分子或化学基团,包括单电荷和多电荷物质。阳离子可为单个原子诸如金属(非限制性示例包括na+或ca+2)、单个分子(非限制性示例包括(ch3)4n+)或化学基团(非限制性示例包括-n(ch3)3+)。术语“胺阳离子”是指nr4+形式的特定分子阳离子,其中四个取代的r部分可独立地选自h和烷基,非限制性示例包括nh4+(铵)、ch3nh3+(甲基铵)、ch3ch2nh3+(乙基铵)、(ch3)2nh2+(二甲基铵)、(ch3)3nh+(三甲基铵)和(ch3)4n+(四甲基铵)。
术语“阴离子”是指具有净负电荷的原子、分子或化学基团,包括单电荷和多电荷物质。阴离子可为单个原子(例如但不限于卤素f-、cl-、br-)、单个分子(非限制性示例包括co3-2、h2po4-、hpo4-2、po4-3、hso4-、so4-2)或化学基团(非限制性示例包括硫酸根、磷酸根、磺酸根、膦酸根、次膦酸根、磺酸根、巯基、羧酸根、氧化胺、异羟肟酸根和羟氨基)。如果移除质子得到净负电荷,则先前定义的化学基团的去质子化形式被认为是阴离子基团。在溶液中,根据henderson-hasselbach公式(ph=pka+log10([a-]/[ha];其中[ha]为未解离酸的摩尔浓度,并且[a-]为该酸的共轭碱的摩尔浓度),化学基团能够失去一个质子而变成作为ph的函数的阴离子。当溶液的ph等于官能团的pka值时,50%的官能团将为阴离子,而剩余的50%将具有质子。通常,如果ph处于或高于官能团的pka,则溶液中的官能团可被认为是阴离子。
术语“盐”是指一种或多种阴离子和阳离子的电中性组合。例如,当r表示为羧酸根基团的盐-coor时,应当理解,羧酸根(-coo-)为具有负电荷-1的阴离子,并且r为具有正电荷+1以与具有电荷-1的一个阴离子形成电中性实体的阳离子,或者r为具有正电荷+2以与均具有电荷-1的两个阴离子形成电中性实体的阳离子。
如本文所用,术语“饱和”是指如此修饰的化学化合物或基团不具有碳-碳双键并且不具有碳-碳三键,除非如下所述。就饱和化学基团的取代型式而言,可存在一个或多个碳氧双键或碳氮双键。当存在此类键时,则不排除碳-碳双键可作为酮-烯醇互变异构或亚胺/烯胺互变异构的一部分出现。
术语“脂族”在没有修饰词“取代”的情况下使用时表示如此修饰的化学化合物/基团是无环或环状的,但为非芳烃化学化合物或基团。在脂族化学化合物/基团中,碳原子可以直链、支链或非芳环(脂环烃)形式连接在一起。脂族化学化合物/基团可以为饱和的,即通过单键(烷烃/烷基)连接;也可以为不饱和的,即具有一个或多个双键(链烯/链烯基)或具有一个或多个三键(炔烃/炔基)。
术语“烷基”在没有修饰词“取代”的情况下使用时是指以碳原子作为连接点的一价饱和脂族基团,其具有直链或支链、环、环状或无环结构,并且不具有除碳和氢之外的原子。因此,如本文所用,环烷基是烷基的子集,其中形成连接点的碳原子也是一个或多个非芳族环结构的成员,其中环烷基基团不包含除碳和氢之外的原子。如本文所用,该术语不排除连接到该环或环系的(碳数限制允许的)一个或多个烷基基团的存在。基团-ch3(me)、-ch2ch3(et)、-ch2ch2ch3(n-pr或丙基)、-ch(ch3)2(i-pr、'pr或异丙基)、-ch(ch2)2(环丙基)、-ch2ch2ch2ch3(n-bu)、-ch(ch3)ch2ch3(仲丁基)、-ch2ch(ch3)2(异丁基)、-c(ch3)3(叔丁基、t-butyl、t-bu或tbu)、-ch2c(ch3)3(新戊基)、环丁基、环戊基、环己基和环己基甲基为烷基基团的非限制性示例。术语“链烷二基”在没有修饰词“取代”的情况下使用时是指以一个或两个饱和碳原子作为连接点的二价饱和脂族基团,其具有直链或支链、环、环状或无环结构,不具有碳-碳双键或三键,并且不具有除碳和氢之外的原子。基团-ch2(亚甲基)、-ch2ch2-、-ch2c(ch3)2ch2-和-ch2ch2ch2-为链烷二基基团的非限制性示例。术语“烷叉基”在没有修饰词“取代”的情况下使用时是指二价基团=crr',其中r和r’独立地为氢、烷基,或者r和r’合在一起表示具有至少两个碳原子的链烷二基。烷叉基基团的非限制性示例包括:=ch2、=ch(ch2ch3)和=c(ch3)2。“烷烃”是指化合物h-r,其中如上对该术语的定义,r为烷基。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3、-s(o)2nh2、-p(o)(oh)2、-p(o)(oh)op(o)(oh)2、-op(o)(oh)2、-op(o)(oh)op(o)(oh)2、-s(o)2(oh)或-os(o)2(oh)取代。以下基团为取代的烷基基团的非限制性示例:-ch2oh、-ch2cl、-cf3、-ch2cn、-ch2c(o)oh、-ch2c(o)och3、-ch2c(o)nh2、-ch2c(o)ch3、-ch2och3、-ch2oc(o)ch3、-ch2nh2、-ch2n(ch3)2、-ch2ch2cl、-ch2p(o)(oh)2、-ch2p(o)(oh)op(o)(oh)2、-ch2s(o)2(oh)和-ch2os(o)2(oh)。术语“卤代烷基”为取代的烷基的子集,其中一个或多个氢原子已被卤素基团取代,并且不存在除碳、氢和卤素之外的其他原子。基团-ch2cl为卤代烷基的非限制性示例。术语“氟代烷基”为取代的烷基的子集,其中一个或多个氢已被氟基团取代,并且不存在除碳、氢和氟之外的其他原子。基团-ch2f、-cf3和-ch2cf3为氟代烷基基团的非限制性示例。
术语“膦酰基烷基”为取代的烷基的子集,其中一个或多个氢已被膦酸根基团取代,并且不存在除碳、氢、磷和氧之外的其他原子。基团-ch2p(o)(oh)2和-ch2ch2p(o)(oh)2以及它们对应的去质子化形式为膦酰基烷基的非限制性示例。
术语“膦酰基(磷酸)烷基”为取代的烷基的子集,其中一个或多个氢已被膦酰基-磷酸根基团取代,并且不存在除碳、氢、磷和氧之外的其他原子。基团-ch2p(o)(oh)op(o)(oh)2和-ch2ch2p(o)(oh)op(o)(oh)2以及它们对应的去质子化形式为膦酰基(磷酸)烷基的非限制性示例。
术语“磺酰基烷基”为取代的烷基的子集,其中一个或多个氢已被磺酸根基团取代,并且不存在除碳、氢、硫和氧之外的其他原子。基团-ch2s(o)2oh和-ch2ch2s(o)2oh以及它们对应的去质子化形式为磺酰基烷基的非限制性示例。
术语“链烯基”在没有修饰词“取代”的情况下使用时是指具有一个碳原子作为连接点的一价不饱和脂族基团,其具有直链或支链、环、环状或无环结构、至少一个非芳族碳-碳双键,不具有碳-碳三键并且不具有除碳和氢之外的原子。链烯基基团的非限制性示例包括:-ch=ch2(乙烯基)、-c(ch3)=ch2(甲基乙烯基)、-ch=chch3、-ch=chch2ch3、-ch2ch=ch2(烯丙基)、-ch2ch=chch3和-ch=chch=ch2。术语“链烯二基”在没有修饰词“取代”的情况下使用时是指具有两个碳原子作为连接点的二价不饱和脂族基团,其具有直链或支链、环、环状或无环结构、至少一个非芳族碳-碳双键,不具有碳-碳三键并且不具有除碳和氢之外的原子。基团>c=ch2(亚乙烯基)、-ch=ch-、-ch=c(ch3)ch2-和-ch=chch2-是链烯二基基团的非限制性示例。需注意,虽然链烯二基基团为脂族,但一旦在两个末端处连接,则不排除该基团形成芳族结构的一部分。术语“链烯”或“烯烃”是同义的,并且是指具有式h-r的化合物,其中如上对该术语的定义,r为烯基。“末端链烯”是指具有仅一个碳-碳双键的链烯,其中该键在分子的一端形成乙烯基基团。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。基团-ch=chf、-ch=chcl和-ch=chbr是取代的链烯基基团的非限制性示例。
术语“炔基”在没有修饰词“取代”的情况下使用时是指具有一个碳原子作为连接点的一价不饱和脂族基团,其具有直链或支链、环、环状或无环结构、至少一个碳-碳三键,并且不具有除碳和氢之外的原子。如本文所用,术语炔基不排除一个或多个非芳族碳-碳双键的存在。基团-c≡ch、-c≡cch3和-ch2c≡cch3是炔基基团的非限制性示例。“炔烃”是指化合物h-r,其中r为炔基。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。
术语“芳基”在没有修饰词“取代”的情况下使用时是指具有一个芳族碳原子作为连接点的一价不饱和芳族基团,所述碳原子形成一个或多个六元芳族环结构的一部分,其中环原子全部为碳,并且其中基团不包含除碳和氢之外的原子。如果存在多于一个环,则环可以是稠合的或未稠合的。如本文所用,该术语不排除连接到第一芳族环或存在的任何另外的芳族环的(碳数限制允许的)一个或多个烷基或芳烷基基团的存在。芳基基团的非限制性示例包括苯基(-ph)、甲基苯基、(二甲基)苯基、-c6h4ch2ch3(乙基苯基)、萘基和衍生于联苯基的一价基团。术语“芳二基”在没有修饰词“取代”的情况下使用时是指具有两个芳族碳原子作为连接点的二价芳族基团,所述碳原子形成一个或多个六元芳族环结构的一部分,其中环原子全部为碳,并且其中一价基团不包含除碳和氢之外的原子。如本文所用,该术语不排除连接到第一芳族环或存在的任何另外的芳族环的(碳数限制允许的)一个或多个烷基、芳基或芳烷基基团的存在。如果存在多于一个环,则环可以是稠合的或未稠合的。未稠合的环可经由(碳数限制允许的)以下中的一者或多者连接:共价键、链烷二基或链烯二基基团。芳二基基团的非限制性示例包括:
“芳烃”是指化合物h-r,其中如上对该术语的定义,r为芳基。苯和甲苯为芳烃的非限制性示例。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。
术语“酰基”在没有修饰词“取代”的情况下使用时是指基团-c(o)r,其中如上文对那些术语的定义,r为氢、烷基、芳基、芳烷基或杂芳基。基团-cho(甲酰基)、-c(o)ch3(乙酰基,ac)、-c(o)ch2ch3、-c(o)ch2ch2ch3、-c(o)ch(ch3)2、-c(o)ch(ch2)2、-c(o)c6h5、-c(o)c6h4ch3、-c(o)ch2c6h5、-c(o)(咪唑基)为酰基基团的非限制性示例。以类似的方式定义“硫代酰基”,不同的是基团-c(o)r的氧原子已被硫原子取代成为-c(s)r。如上所定义,术语“醛”对应于烷烃,其中氢原子中的至少一个已被-cho基团取代。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子(包括直接连接羰基或硫代羰基基团的氢原子,如果有的话)已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。基团-c(o)ch2cf3、-co2h(羧基)、-co2ch3(甲基羧基)、-co2ch2ch3、-c(o)nh2(氨甲酰基)和-con(ch3)2为取代的酰基的非限制性示例。
术语“烷氧基”在没有修饰词“取代”的情况下使用时是指基团-or,其中如上文对该术语的定义,r为烷基。烷氧基基团的非限制性示例包括:-och3(甲氧基)、-och2ch3(乙氧基)、-och2ch2ch3、-och(ch3)2(异丙氧基)、-o(ch3)3(叔丁氧基)、-och(ch2)2、-o-环戊基和-o-环己基。术语“烯氧基”、“炔氧基”、“芳氧基”、“芳烷氧基”、“杂芳氧基”、“杂环烷氧基”和“酰氧基”在没有修饰词“取代”的情况下使用时是指定义为-or的基团,其中r分别为烯基、炔基、芳基、芳烷基、杂芳基、杂环烷基和酰基。术语“烷氧基二基”是指二价基团-o-链烷二基-、-o-链烷二基-o-、或-链烷二基-o-链烷二基-。术语“链烷二基-烷氧基”是指-链烷二基-o-烷基。链烷二基-烷氧基的非限制性示例为-ch2-ch2-o-ch2-ch3。术语“烷硫基”和“酰硫基”在没有修饰词“取代”的情况下使用时是指基团-sr,其中r分别为烷基和酰基。术语“醇”对应于如上定义的烷烃,其中至少一个氢原子已被羟基基团取代。术语“醚”对应于如上定义的烷烃,其中至少一个氢原子已被烷氧基基团取代。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。
术语“烷基氨基”在没有修饰词“取代”的情况下使用时是指基团-nhr,其中如上文对该术语的定义,r为烷基。烷基氨基基团的非限制性示例包括:-nhch3和-nhch2ch3。术语“二烷基氨基”在没有修饰词“取代”的情况下使用时是指基团-nrr',其中r和r'可以是相同或不同的烷基,或者r和r'可以合在一起表示链烷二基。二烷基氨基基团的非限制性示例包括:-n(ch3)2、-n(ch3)(ch2ch3)和n-吡咯烷基。术语“烷氧基氨基”、“烯基氨基”、“炔基氨基”、“芳氨基”、“芳烷基氨基”、“杂芳氨基”、“杂环烷基氨基”和“烷基磺酰基氨基”在没有修饰词“取代”的情况下使用时是指定义为-nhr的基团,其中r分别为烷氧基、烯基、炔基、芳基、芳烷基、杂芳基、杂环烷基和烷基磺酰基。芳氨基基团的非限制性示例为-nhc6h5。术语“酰氨基(酰基氨基)”在没有修饰词“取代”的情况下使用时是指基团-nhr,其中如上文对该术语的定义,r为酰基。酰氨基基团的非限制性示例为-nhc(o)ch3。术语“烷基亚氨基”在没有修饰词“取代”的情况下使用时是指二价基团=nr,其中如上文对该术语的定义,r为烷基。术语“烷基氨基二基”是指二价基团-nh-链烷二基-、-nh-链烷二基-nh-、或-链烷二基-nh-链烷二基-。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。基团-nhc(o)och3和-nhc(o)nhch3为取代的酰氨基基团的非限制性示例。
术语“烷基磺酰基”和“烷基亚磺酰基”在没有修饰词“取代”的情况下使用时分别是指基团-s(o)2r和-s(o)r,其中如上文对该术语的定义,r为烷基。以类似的方式定义术语“烯基磺酰基”、“炔基磺酰基”、“芳基磺酰基”、“芳烷基磺酰基”、“杂芳基磺酰基”和“杂环烷基磺酰基”。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。
术语“烷基磷酸根”在没有修饰词“取代”的情况下使用时是指基团-op(o)(oh)(or)或其去质子化形式,其中如上文对该术语的定义,r为烷基。烷基磷酸根基团的非限制性示例包括:-op(o)(oh)(ome)和-op(o)(oh)(oet)。术语“二烷基磷酸根”在没有修饰词“取代”的情况下使用时是指基团-op(o)(or)(or'),其中r和r'可以是相同或不同的烷基,或者r和r'可以合在一起表示链烷二基。二烷基磷酸根基团的非限制性示例包括:-op(o)(ome)2、-op(o)(oet)(ome)和-op(o)(oet)2。当这些术语中的任一个与修饰词“取代”一起使用时,一个或多个氢原子已独立地被-oh、-f、-cl、-br、-i、-nh2、-no2、-co2h、-co2ch3、-cn、-sh、-och3、-och2ch3、-c(o)ch3、-nhch3、-nhch2ch3、-n(ch3)2、-c(o)nh2、-oc(o)ch3或-s(o)2nh2取代。
连接基团是指两个其他限定基团之间的共价键,或连接两个其他限定基团的一系列共价键合的原子,其中在该一系列共价键合的原子中,除了连接到两个其他限定基团的位点以外,不具有开放价态。连接基团的非限制性示例包括共价键、链烷二基、链烯二基、芳二基、烷氧基二基和烷基氨基二基。
如本文所用,“手性助剂”是指能够影响反应的立体选择性的可移除手性基团。本领域的技术人员熟悉此类化合物,并且许多此类化合物是可商购获得的。
本文所用的其他缩写如下:dmso,二甲基亚砜;dmf,二甲基甲酰胺;mecn,乙腈;meoh,甲醇;etoh,乙醇;etoac,乙酸乙酯;tbuoh,叔丁醇;iproh,异丙醇;chexoh,环己醇;ac2o,乙酸酐;acooh,过乙酸;hco2et,甲酸乙酯;thf,四氢呋喃;mtbe,甲基叔丁基醚;dme,二甲氧基乙烷;nbs,n-溴代琥珀酰亚胺;cdi,羰基二咪唑;diea,二异丙基乙胺;tea,三乙胺;dmap,二甲基氨基吡啶;naoh,氢氧化钠;aaph,2,2'-偶氮双(2-甲基丙脒)二盐酸盐;cta,1-辛硫醇;aps,过硫酸铵;tmp,磷酸三甲酯;vpa,乙烯基膦酸;vpp,乙烯基膦酰基-单磷酸盐;vppp,乙烯基膦酰基-焦磷酸盐;mvpp,甲基-乙烯基膦酰基-单磷酸盐;svs,乙烯基磺酸钠;amps,2-丙烯酰氨基-2-甲基丙烷磺酸钠;spa,3-磺丙基丙烯酸酯的钾盐;22a2mpa2hcl,2,2'-偶氮双(2-甲基丙脒)二盐酸盐;vbpp,(4-乙烯基苄基)单膦酰基-磷酸盐;vsme,乙烯基磺酸甲酯;naome,甲醇钠;nacl,氯化钠;dmvp,二甲基乙烯基膦酸盐
国际纯粹与应用化学联合会(iupac)将“单体分子”定义为“可经历聚合从而为大分子基本结构贡献结构单元的分子”。聚合物为大分子。
iupac将“聚合物主链”或“主链”定义为“所有其他链(长链或短链或这两者)可视为其侧基的线性链”,需注意,“在两条或多条链可等同地视为主链的情况下,选择使得分子表示最简单的链。”根据制备主链的原料,主链可具有不同的化学组成。来自化学合成和生物合成的聚合物的常见主链包括烷烃(通常来自乙烯基或甲基乙烯基聚合或阳离子和阴离子聚合)、聚酯(来自缩聚)、聚酰胺(诸如来自涉及酰胺化反应的聚合的多肽)以及来自环氧化物开环的聚乙氧基化物。
iupac将“侧基”或“侧部基团”定义为“来自主链的支链,既非低聚物也非聚合物”。此类侧基不包括线性重复单元。
iupac将“聚合物侧链”或“侧链”定义为“来自大分子链的低聚或聚合支链”,另外需注意,“低聚支链可被称为短支链”并且“聚合物支链可被称为长支链”。
将“后聚合改性”定义为在聚合后发生的聚合物的任何反应或处理。后聚合改性包括对聚合物主链、侧基或聚合物侧链内或连接到聚合物主链、侧基或聚合物侧链的化学基团的反应。
当与权利要求和/或说明书中的术语“包含”一起使用时,词语“一个”的使用可意指“一个”,但其也与“一个或多个”、“至少一个”和“一个或多于一个”的含义一致。
在整个本申请中,术语“约”用于指示值包括了用于确定该值的设备、方法误差的固有变化或研究对象之间存在的变化。
术语“包含”、“具有”和“包括”是开放式连接动词。这些动词中的一个或多个的任何形式或时态,诸如“包含”、“具有”和“包括”也是开放式的。例如,“包含”、“具有”或“包括”一个或多个步骤的任何方法不限于拥有仅那些一个或多个步骤并且还涵盖其他未列出的步骤。
以上定义代替了以引用方式并入本文的任何参考文献中的任何冲突定义。然而,某些术语被定义的事实不应被视为指示任何未定义的术语是不确定的。相反,所用的所有术语都被认为是以术语描述本发明,使得普通技术人员可理解本发明的范围并实践本发明。
含有膦酰基-磷酸根的聚合物
在一个实施方案中,本发明涉及具有式1的结构的化合物:
其中:
r1选自-h和-ch3;
r2选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式2的结构:
其中:
δ为与式1的连接位点,
r5和r6独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;
r3选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式3的结构:
其中:
δ为与式1的连接位点,
r7和r8独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
n为1至22的整数;
r4选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
l选自化学键、芳二基和式4的结构:
其中:
α为与链烯基基团的连接位点;
β为与膦酰基-磷酸根基团的连接位点;
x选自式5至式11所示的结构;
其中:
r9选自-h、烷基(c1-8)、膦酰基烷基和膦酰基(磷酸)烷基;并且
y选自链烷二基、烷氧基二基、烷基氨基二基和链烯二基。
在化合物的一个实施方案中,式1的r1为h。在化合物的另一个实施方案中,式1的r1为ch3。在化合物的另一个实施方案中,式1的l为共价键。
在化合物的一个实施方案中,r2、r3和r4独立地选自h、na盐和k盐。在化合物的一个实施方案中,r2、r3和r4独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。
在化合物的另一个实施方案中,r2具有式2的结构。在另一个实施方案中,r2具有式2的结构,并且r5和r6独立地选自h、na盐和k盐。在另一个实施方案中,r2具有式2的结构,并且r5和r6独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。
在化合物的另一个实施方案中,r3具有式3的结构。在化合物的另一个实施方案中,r3具有式3的结构,并且n为1至3的整数。在化合物的另一个实施方案中,r3具有式3的结构,并且n为1。在化合物的另一个实施方案中,r3具有式3的结构,并且r7和r8独立地选自h、na盐和k盐。在化合物的另一个实施方案中,r3具有式3的结构,并且r7和r8独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。在化合物的另一个实施方案中,r3具有式3的结构,r7和r8独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐,并且n为1。
在一个实施方案中,r1为h,并且l为共价键。在另一个实施方案中,r1为ch3,并且l为共价键。在另一个实施方案中,r1为h,l为共价键,并且r2、r3和r4独立地选自h、na盐和k盐。在另一个实施方案中,r1为ch3,l为共价键,并且r2、r3和r4独立地选自h、na盐和k盐。在另一个实施方案中,r1为h,l为共价键,并且r2具有式2的结构。在另一个实施方案中,r1为ch3,l为共价键,并且r2具有式2的结构。在另一个实施方案中,r1为h,l为共价键,并且r3具有式3的结构。在另一个实施方案中,r1为ch3,l为共价键,并且r3具有式3的结构。
在化合物的一个实施方案中,l具有式4的结构,并且x具有式5的结构。在化合物的另一个实施方案中,l具有式4的结构,并且x具有式8的结构。在化合物的一个实施方案中,l具有式4的结构,并且x具有式10的结构。在化合物的一个实施方案中,l具有式4的结构,并且x具有式6的结构。在化合物的另一个实施方案中,l具有式4的结构,x具有式5的结构,并且y为链烷二基。在化合物的另一个实施方案中,l具有式4的结构,x具有式8的结构,并且y为链烷二基。在化合物的一个实施方案中,l具有式4的结构,x具有式10的结构,并且y为链烷二基。在化合物的一个实施方案中,l具有式4的结构,x具有式6的结构,并且y选自链烷二基和烷氧基二基。
本发明的另一个实施方案为一种新型聚合物。该聚合物包含膦酰基-磷酸根基团,其中该膦酰基-磷酸根基团具有式12的结构:
其中:
ε为与所述聚合物的主链、侧基或侧链中的碳原子的连接位点;
r10选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式13的结构:
其中:
θ为与式12的连接位点,
r13和r14独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;
r11选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐和胺阳离子盐,以及式14的结构:
其中:
θ为与式12的连接位点,
r15和r16独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
n为1至22的整数;并且
r12选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐。
在聚合物的一个实施方案中,r10、r11和r12独立地选自h、na盐和k盐。在聚合物的一个实施方案中,r10、r11和r12独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。
在聚合物的另一个实施方案中,r10具有式13的结构。在另一个实施方案中,r10具有式3的结构,并且r13和r14独立地选自h、na盐和k盐。在另一个实施方案中,r10具有式13的结构,并且r13和r14独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。
在聚合物的另一个实施方案中,r11具有式14的结构。在聚合物的另一个实施方案中,r11具有式14的结构,并且n为1至3的整数。在聚合物的另一个实施方案中,r11具有式14的结构,并且n为1。在聚合物的另一个实施方案中,r11具有式14的结构,并且r15和r16独立地选自h、na盐和k盐。在聚合物的另一个实施方案中,r11具有式14的结构,并且r15和r16独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。在聚合物的另一个实施方案中,r11具有式14的结构,r15和r16独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐,并且n为1。
在一个实施方案中,用于形成该聚合物的至少一种单体包含膦酰基-磷酸根基团。在另一个实施方案中,在后聚合改性过程中加入该膦酰基-磷酸根基团。
在另一个实施方案中,用于形成该聚合物的至少一种单体具有式15的结构:
其中:
ω为与式12的膦酰基-磷酸根基团的连接位点;
r17选自-h和-ch3;
l1选自化学键、芳二基和式4的结构:
其中:
α为与链烯基基团的连接位点;
β为与式12的膦酰基-磷酸根基团的连接位点;
x选自式5至式11中的结构;
其中:
r9选自-h、烷基(c1-8)、膦酰基烷基和膦酰基(磷酸)烷基;并且
y选自链烷二基、烷氧基二基、烷基氨基二基和链烯二基。
在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为h。在另一个实施方案中,当用于形成聚合物的至少一种单体具有式15的结构时,r17为ch3。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1为共价键。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为h,并且l1为共价键。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为ch3,并且l1为共价键。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r10、r11和r12独立地选自-h、na盐和k盐。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为h,l1为共价键,并且r10、r11和r12独立地选自-h、na盐和k盐。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为ch3,l1为共价键,并且r10、r11和r12独立地选自h、na盐和k盐。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为h,l1为共价键,并且r10具有式13的结构。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为ch3,l1为共价键,并且r10具有式13的结构。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为h,l1为共价键,并且r11具有式14的结构。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,r17为ch3,l1为共价键,并且r11具有式14的结构。
在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式5的结构。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式8的结构。在一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式10的结构。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构并且x具有式6的结构。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构,x具有式5的结构,并且y为链烷二基。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构,x具有式8的结构,并且y为链烷二基。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构,x具有式10的结构,并且y为链烷二基。在另一个实施方案中,当用于形成该聚合物的至少一种单体具有式15的结构时,l1具有式4的结构,x具有式6的结构,并且y选自链烷二基和烷氧基二基。
本发明的另一个实施方案是新型聚合物,在此上下文中该聚合物旨在包括低聚物诸如二聚体、三聚体和四聚体。该聚合物包含膦酰基-磷酸根基团,其中该膦酰基-磷酸根基团具有式16的结构:
其中:
r1选自-h和-ch3;
r2选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式2的结构:
其中:
δ为与式16的连接位点,
r5和r6独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;
r3选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐、胺阳离子盐和式3的结构:
其中:
δ为与式16的连接位点,
r7和r8独立地选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;并且
n为1至22的整数;
r4选自-h、烷基、链烷二基烷氧基、具有na、k、ca、mg、mn、zn、fe或sn阳离子的金属盐以及胺阳离子盐;
r18为由聚合物引发产生的化学基团;
r19为导致链终止的化学基团;
m2选自化学键和一种或多种共聚单体的后聚合残基;
m为2至450的整数;并且
l选自化学键、芳二基和式4的结构:
其中:
α为与链烯基基团的连接位点;
β为与膦酰基-磷酸根基团的连接位点;
x选自式5至式11所示的结构;
其中:
r9选自-h、烷基(c1-8)、膦酰基烷基和膦酰基(磷酸)烷基;并且
y选自链烷二基、烷氧基二基、烷基氨基二基和链烯二基。
在聚合物的一个实施方案中,式16的r1为h。在聚合物的另一个实施方案中,式16的r1为ch3。在聚合物的另一个实施方案中,式16的l为共价键。
在聚合物的一个实施方案中,r2、r3和r4独立地选自h、na盐和k盐。在聚合物的一个实施方案中,r2、r3和r4独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。
在聚合物的另一个实施方案中,r2具有式2的结构。在另一个实施方案中,r2具有式2的结构,并且r5和r6独立地选自h、na盐和k盐。在另一个实施方案中,r2具有式2的结构,并且r5和r6独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。
在聚合物的另一个实施方案中,r3具有式3的结构。在聚合物的另一个实施方案中,r3具有式3的结构,并且n为1至3的整数。在聚合物的另一个实施方案中,r3具有式3的结构,并且n为1。在聚合物的另一个实施方案中,r3具有式3的结构,并且r7和r8独立地选自h、na盐和k盐。在聚合物的另一个实施方案中,r3具有式3的结构,并且r7和r8独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐。在聚合物的另一个实施方案中,r3具有式3的结构,r7和r8独立地选自h、na盐、k盐、zn盐、ca盐、sn盐和胺阳离子盐,并且n为1。
在一个实施方案中,r1为h,并且l为共价键。在另一个实施方案中,r1为ch3,并且l为共价键。在另一个实施方案中,r1为h,l为共价键,并且r2、r3和r4独立地选自h、na盐、k盐和胺阳离子盐。在另一个实施方案中,r1为ch3,l为共价键,并且r2、r3和r4独立地选自h、na盐、k盐和胺阳离子盐。在另一个实施方案中,r1为h,l为共价键,并且r2具有式2的结构。在另一个实施方案中,r1为ch3,l为共价键,并且r2具有式2的结构。在另一个实施方案中,r1为h,l为共价键,并且r3具有式3的结构。在另一个实施方案中,r1为ch3,l为共价键,并且r3具有式3的结构。
在聚合物的一个实施方案中,l具有式4的结构,并且x具有式5的结构。在聚合物的另一个实施方案中,l具有式4的结构,并且x具有式8的结构。在聚合物的一个实施方案中,l具有式4的结构,并且x具有式10的结构。在聚合物的一个实施方案中,l具有式4的结构,并且x具有式6的结构。在聚合物的另一个实施方案中,l具有式4的结构,x具有式5的结构,并且y为链烷二基。在聚合物的另一个实施方案中,l具有式4的结构,x具有式8的结构,并且y为链烷二基。在聚合物的一个实施方案中,l具有式4的结构,x具有式10的结构,并且y为链烷二基。在聚合物的一个实施方案中,l具有式4的结构,x具有式6的结构,并且y选自链烷二基和烷氧基二基。
在聚合物的一个实施方案中,得自聚合物引发的化学基团r18选自式17至式21的结构:
其中:
r20选自-h、na、k和胺阳离子盐;
τ为与聚合物主链的连接位点;并且
q为用于聚合的单体的非烯烃残基。
在另一个实施方案中,q具有式22的结构:
其中:
l、r2、r3和r4如前所述,并且κ表示与式21的连接位点。
在聚合物的一个实施方案中,m2为具有式23的结构的一种或多种共聚单体的聚合残基:
其中:
r21选自-h或-ch3;
q1为用于聚合的共聚单体的非烯烃残基;并且
p为1至450的整数。
在另一个实施方案中,q为膦酰基-磷酸根。
在聚合物的一个实施方案中,由聚合物终止产生的化学基团r19选自-h。在聚合物的一个实施方案中,由聚合物终止产生的化学基团r19为具有头对头连接的另一聚合物链。
在聚合物的一个优选的实施方案中,r1为h,l为共价键,并且r2、r3和r4独立地选自h、na盐、k盐和胺阳离子盐,r18为式21的结构,q为图22的结构,并且r19为h。
制备聚合物的方法
本发明的实施方案可使用以下这些通用方法来制得。
本发明的聚合物可由多种技术制得,包括本体聚合反应、溶液聚合反应、乳液聚合反应或悬浮聚合反应。聚合方法和用于聚合的技术概述于encyclopediaofpolymerscienceandtechnology(intersciencepublishers,newyork)第7卷第361-431页(1967),和kirk-othmerencyclopediaofchemicaltechnology第3版第18卷第740-744页(johnwiley&sons,newyork,1982)中,将两篇文献均以引用方式并入本文。适用于本发明的一般反应技术还参见sorenson,w.p.和campbell,t.w.的preparativemethodsofpolymerchemistry.第2版(intersciencepublishers,newyork,1968)第248-251页,将所述文献以引用方式并入本文。在一个例子中,使用水溶性引发剂,由自由基共聚反应制得所述聚合物。适宜的自由基引发剂包括但不限于热引发剂、氧化还原对和光化学引发剂。氧化还原和光化学引发剂可用于在低于约30℃的温度下引发的聚合过程。此类引发剂概述于kirk-othmerencyclopediaofchemicaltechnology第3版(johnwiley&sons,newyork)第13卷第355-373页(1981)中,将所述文献以引用方式并入本文。可在30℃或更低的温度下提供自由基的典型水溶性引发剂包括氧化还原对,如过硫酸钾/硝酸银,和抗坏血酸/过氧化氢。在一个示例中,在高于40℃下实施的聚合过程中,该方法使用热引发剂。可使用可在40℃或更高的温度下提供自由基的水溶性引发剂。这些包括但不限于过氧化氢、过硫酸铵、和2,2'-偶氮二(2-脒基丙烷)二盐酸盐。在一个示例中,使用过硫酸铵为引发剂,水溶性起始单体在60℃的水中聚合。
线型聚合物的末端处的化学官能团的种类取决于该聚合物链的聚合如何引发和终止。对于自由基聚合,体系中的任何自由基都可开始新链。该自由基可为引发剂的直接衍生物,诸如来自过硫酸盐的硫酸根基团,或来自偶氮型引发剂的烷基基团(诸如但不限于2,2'偶氮二(2-脒基丙烷)二盐酸盐)。自由基也可为转移反应的结果,例如在水和另一个自由基之间以产生羟基自由基或在磷酸根和另一个自由基之间以产生磷酸根自由基。下面给出了这些所得结构的非限制性示例,其中r表示h或适当的抗衡离子诸如na、k或胺,并且τ表示与聚合物的连接位点。
自由基也可为链转移反应的结果,其中自由基从增长的聚合物链转移以开始新链。在乙烯基膦酸盐单体的聚合反应中已明确指出链转移。
在含膦酰基-磷酸根的聚合物中,在最终的聚合物组合物中观察到乙烯基ch2基团。假设这些乙烯基基团由两种机制中的一种形成。第一机制是与
引入乙烯基基团的第二机制涉及回咬反应和β断裂。该机制在文献中已被广泛指出用于丙烯酸酯聚合物。β断裂后产生乙烯基基团和伯自由基。
使用先前所用的利用τ来表示与聚合物的连接位点的命名,初始官能团可写做如下。应该指出的是,链转移和回咬以及随后的β断裂机制将产生在同一碳原子上具有两个质子的乙烯基基团。
聚合物链的末端上的化学基团取决于链是如何终止的。最常见的终止是前面提到的链转移和回咬反应以及组合和歧化。在链转移和反向破坏中,末端基团通常为氢。在组合中,两个链上的链传播自由基反应以形成新链。该反应在连接点处引起“头对头”构型。
在歧化中,氢从一个自由基链交换到另一个自由基链。结果是一条链是不饱和的,而另一条链是饱和的。需注意,所得的不饱和基团不是乙烯基基团。不饱和基团中的每个碳仅具有一个氢。
包含膦酰基-磷酸根基团的聚合物可具有直接连接在聚合物侧基或侧链而不在聚合物主链上的膦酰基-磷酸根基团。可通过具有膦酰基-磷酸根基团的单体的聚合反应,或者通过不具有膦酰基-磷酸根基团的单体的聚合反应以及随后的所得聚合物的后聚合改性来添加膦酰基-磷酸根基团,使该膦酰基-磷酸根基团结合到聚合物中。虽然为了简单起见后续段落中的示例将示出均聚物,但应当理解,具有除膦酸根或含膦酰基-磷酸根的单体之外的另外单体的聚合物可通过在聚合过程中包括这些其他单体来制备。
作为包含连接到聚合物主链上的膦酰基-磷酸根基团的聚合物的示例,考虑由单体乙烯基膦酸盐或甲基-乙烯基膦酸盐制成的聚合物。可使乙烯基膦酸盐或甲基-乙烯基膦酸盐化学反应以形成膦酰基-磷酸盐单体,如方案1中的反应1所示。然后这些含膦酰基-磷酸根的单体可聚合,如相同方案的反应2所示,以得到具有直接连接到聚合物主链的膦酰基-磷酸根基团的含膦酰基-磷酸根的聚合物。另选地,可首先如反应3所示使乙烯基膦酸盐或甲基-乙烯基膦酸盐聚合以得到聚合物。在聚合之后,膦酰基-磷酸根基团可通过如反应4所示使连接的膦酸盐部分反应进行后聚合改性而产生,从而产生直接连接到聚合物主链的膦酰基-磷酸根基团。
通过后聚合改性产生直接连接到主链的膦酰基-磷酸根基团的第二种方式可通过从聚乙烯开始举例说明。对于此类修饰中的第一反应的示例,参见m.anbar,g.a.st.john和a.cscott,jdentres,第53卷,第4期,第867-878页,1974年。如方案2所示,聚乙烯首先用氧和pcl3进行氧化磷酸化以形成无规膦酸化聚合物。然后可将该膦酸化聚合物改性以产生无规取代的膦酰基-磷酸盐聚合物。所示反应产物旨在示出所得聚合物中膦酰基-磷酸根基团连接点的无规性质。
作为制备具有连接到侧基的膦酰基-磷酸根基团的聚合物的示例,考虑方案3中所示的乙烯基苄基化学物质。与先前的示例一样,为简单起见,该方案将示出均聚物。然而,可通过在聚合过程中包括另外的单体来制备具有另外的单体成分的杂聚物。4-乙烯基苄基氯可与二乙基亚磷酸盐反应以形成乙烯基苄基膦酸盐,如方案3的反应1所示。对于该反应的示例,参见frantz,richard;durand,jean-olivier;carre,francis;lanneau,gerardf.;lebideau,jean;alonso,bruno;massiot,dominique,chemistry-aeuropeanjournal,第9卷,第3期,第770-775页,2003年。可使乙烯基苄基膦酸盐反应以形成反应2中所示的乙烯基苄基膦酰基-磷酸盐,如以下实施例部分所述。然后可使该单体聚合,如以下实施例部分所述,以形成由反应5所示的含膦酰基-磷酸根的聚合物,其中膦酰基-磷酸根基团连接到聚合物上的侧基。另选地,第一中间体乙烯基苄基膦酸盐可如反应4所示聚合以制备聚乙烯基苄基膦酸盐。对于该反应的示例,参见m.anbar,g.a.st.john和a.cscott,jdentres,第53卷,第4期,第867-878页,1974年。然后可如反应7中所示,使聚乙烯基苄基膦酸盐反应,以产生含膦酰基-磷酸根的聚合物,其中膦酰基-磷酸根基团通过以下实施例部分所述的后聚合改性连接到聚合物上的侧基。涉及后聚合改性的第二途径也示于相同的方案中。4-乙烯基苄基氯可聚合以提供反应3中所示的聚乙烯基苄基氯。该聚合物可如反应6中所示被膦酸化(例如参见sanghunkim、youngchulpark、guihyunjung和changgicho,macromoleculesresearch,第15卷第6期,第587-597页,2007年),然后使所得的聚乙烯基苄基膦酸盐反应以产生反应7中所示的含膦酰基-磷酸根的聚合物。
作为包含连接到侧链的膦酰基-磷酸根基团的聚合物的第一示例,考虑方案4中所示的聚乙二醇(peg)侧链。含膦酸根的peg链可与丙烯酰氯反应以产生具有peg封端的膦酸根的丙烯酸酯。在反应产生膦酰基-磷酸盐之后,膦酰基-磷酸盐单体可聚合以产生含膦酰基-磷酸根的聚合物,其中膦酰基-磷酸根连接到聚合物的侧链。
作为包含连接到侧链的膦酰基-磷酸根基团的聚合物的第二示例,考虑方案5中所示的聚乙烯醇。羟基可与环氧乙烷反应以产生具有peg侧链的聚合物。侧链上的封端羟基可与乙烯基膦酸根反应,然后反应以形成膦酰基-磷酸根。因此,本示例描述了含膦酰基-磷酸根的聚合物,其中膦酰基-磷酸根连接到聚合物的侧链并经由后聚合改性加入。
所述方案本质上并非旨在穷举性的,而是旨在传达可产生含膦酰基-磷酸根的聚合物的各种方式。这些示例提供了合成的技术细节和含膦酰基-磷酸根基团的聚合物的众多变型,即膦酰基-磷酸根基团直接连接到聚合物主链的聚合物和膦酰基-磷酸根基团连接到侧基的聚合物两者。对于可转化成含膦酰基-磷酸根的单体和聚合物的含膦酸根的单体和聚合物的另外示例,参见sophiemonge、benjamincanniccioni、ghislaindavid和jean-jacquesrobin,rscpolymerchemistryseriesno.11,phosphorus-basedpolymers:fromsynthesistoapplications,由sophiemonge和ghislaindavid编辑,theroyalsocietyofchemistry2014,由royalsocietyofchemistry公布,www.rsc.org。
含膦酰基-磷酸根的聚合物的用途
可将根据本发明的含膦酰基-磷酸根的聚合物结合到多种组合物中。这些组合物包括水性和非水性组合物两者。这些组合物可用于处理牙齿、毛发、身体、织物、纸材、非织物和硬质表面。这些组合物可用于水处理、锅炉处理、处理船壳、油井、电池、烘焙、发酵、陶瓷、塑料稳定剂、玻璃制造、乳酪生产、食品中的缓冲剂、洁齿剂中的研磨剂、肉中的粘合剂、咖啡奶精、防冻剂、油漆液体皂中的分散剂、金属清洁剂、合成橡胶、纺织物和阻燃剂。这些组合物还可用于处理含多价金属阳离子的材料,该多价金属阳离子包括但不限于钙、锡、镁和铁。此类材料的示例包括羟基磷灰石、碳酸钙(无定形、方解石、文石)、磷酸钙、氢氧化钙、碳酸镁、磷酸镁、肥皂残渣(硬脂酸和碳酸盐的钙盐、镁盐和铁盐的混合物)以及硬水污渍。在一个实施方案中,包括含膦酰基-磷酸根的聚合物的组合物是非水性的。在另一个实施方案中,该组合物为水性的。
可将含膦酰基-磷酸根的化合物和聚合物施用到多种基材上。基材的实施方案包括生物材料、织物、非织造材料、纸制品和硬质表面材料。在一个实施方案中,生物材料包括牙齿。在另一个实施方案中,生物材料包括角蛋白,诸如毛发或皮肤。
实施例
以下实施例进一步描述和证明了本发明范围内优选的实施方案。给出这些实施例仅为了说明性目的,不可理解为是对本发明的限制,因为在不脱离本发明的实质和范围的情况下,它们的许多变型也是可能的。成分可由化学名来确定,或换句话讲如下限定。
粉末防污模型(pspm)
粉末防污模型(pspm)是一种筛选技术,其中将羟基磷灰石粉末(hap)用作污渍积聚的基底。该技术的一般目的是示出和定量用于口腔护理的化学试剂的防牙齿变色能力或牙齿变色可能性。羟基磷灰石粉末提供茶生色底物吸附的大表面区域。用漱口液或洁齿剂形式的口腔护理活性物质预处理hap,根据活性物质阻挡或增强这些生色底物与hap表面结合的能力,得到不同程度的污渍积聚。然后,可通过图像分析对污渍的大小进行定量。pspm中涉及的步骤在下文描述。
1.hap预处理
量取200mg至210mg的hap粉末(
2.hap变色
在最终水洗后,将20ml经过滤的茶(每100ml热水中1个lipton茶袋,渗出5分钟,过滤并在50℃下使用)添加到每个粒料中并涡旋30秒以使hap完全悬浮在茶中。将粉末悬浮液以15,000rpm离心15分钟并滗出。将约25ml水加入到管中,涡旋,然后以15,000rpm离心15分钟。滗出液体并且将洗涤循环再重复2次。
3.用于颜色分析的hap制备
将粒料在大约10ml水中涡旋直至完全悬浮,然后在真空下过滤到millipore过滤盘(膜过滤器4.5tm,47mm目录#hawpo4700,milliporecorporation,bedford,mass.)上。使用约等于200mg未经处理、未变色的hap制备对照盘。然后将过滤盘在平坦位置干燥过夜,然后层合。
4.变色的hap的颜色分析
whitelight系统:将hap盘(未经处理的hap对照物和hap处理物)置于稳定的样本夹持器中。使用具有配备有偏振滤光器的镜头的数码相机(产自canoninc.(melville,ny)的相机型号canoneos70d,具有带适配器的nikon55mmmicro-nikkor镜头)来测量颜色。灯系统由dedo灯(型号dlh2)提供,其配备有150瓦、24v灯泡型号(xenophot型号hlx64640),隔开约30cm定位(从光通过其离开的玻璃透镜中一个到另一个的外部圆形表面中心测量),并且以45度角瞄准,使得光路在hap盘上相交。使用具有ultragrab、optimas和giantimaging软件的whitelight执行图像分析。
5.对照物
单一聚合物pspm的常见对照物是作为处理物然后暴露于茶的水,以及不暴露于茶的水。此外,焦磷酸盐和多磷酸盐作为内部对照物运行。
6.结果
计算l*(亮度)、a*(红色(+)/绿色(—))、b*(黄色(—)/蓝色(+))和e(总颜色)的变化如下:
报告结果为相对于阴性对照物的平均δl、δa、δb和/或δe以及防污百分比(al&ae)。
粉末去污模型(psrm)
粉末去污模型(psrm)是一种筛选技术,其中将羟基磷灰石粉末(hap)用作污渍积聚的基底。该技术的目的是示出和定量用于口腔护理的化学试剂的去污特性。羟基磷灰石粉末提供茶生色底物吸附的大表面区域。用漱口液或洁齿剂形式的口腔护理活性物质处理变色的hap,根据活性物质破坏这些生色底物与hap表面结合的能力,获得不同水平的去污效果。然后,可通过图像分析对去污效果的程度进行定量。该模型的试验可在三天内完成。psrm中涉及的步骤在下文中描述。
1.hap变色
通过将10g的hap粉末在200ml的经过滤的茶中搅拌5分钟来制备大批量的茶锈的hap。分入离心管,并且以15,000rpm离心15分钟。通过加入25ml水中洗涤粒料,涡旋,以15,000rpm离心15分钟,然后吸出液体。确保粒料在涡旋期间破碎。重复洗涤。
将离心管置于对流烘箱(55℃至65℃)中过夜以干燥变色的hap。一旦干燥,将变色的hap汇集在一起并用研杵和研钵研磨成细粉。
2.hap处理
量取200mg至210mg的hap粉末(
3.用于颜色分析的hap制备
将粒料在大约10ml水中涡旋直至完全悬浮,然后在真空下过滤到millipore过滤盘(膜过滤器4.5tm,47mm目录#hawpo4700,milliporecorporation,bedford,mass.)上。使用约等于200mg未经处理的变色的hap制备对照盘。然后将过滤盘在平坦位置干燥过夜,然后层合。
4.变色的hap的颜色分析
whitelight系统:将hap盘(未经处理的hap对照物和hap处理物)置于稳定的样本夹持器中。使用具有配备有偏振滤光器的镜头的数码相机(产自canoninc.(melville,ny)的相机型号canoneos70d,具有带适配器的nikon55mmmicro-nikkor镜头)来测量颜色。灯系统由dedo灯(型号dlh2)提供,其配备有150瓦、24v灯泡型号(xenophot型号hlx64640),隔开约30cm定位(从光通过其离开的玻璃透镜中一个到另一个的外部圆形表面中心测量),并且以45度角瞄准,使得光路在hap盘上相交。使用具有ultragrab、optimas和giantimaging软件的whitelight执行图像分析。
5.对照物
单一聚合物psrm的常见对照物是作为处理物然后暴露于茶的水,以及不暴露于茶的水。此外,焦磷酸盐和多磷酸盐作为内部对照物运行。
6.结果
计算l*(亮度)、a*(红色(+)/绿色(—))、b*(黄色(—)/蓝色(+))和e(总颜色)的变化如下:
报告结果为相对于阴性对照物的平均δl、δa、δb和/或δe以及防污百分比(al&ae)。
体外薄膜茶锈模型(iptsm)
牙齿变色是使用氟化亚锡组合物时常见的不可取副作用。由与聚合物矿物质表面活性剂结合的亚锡来更有效地递送亚锡,使得本文所述的经改善的氟化亚锡洁齿剂减少牙齿色斑的形成。通常由亚锡造成的牙齿表面的变色可在临床情况下通过采用色斑指数诸如文献中所述的lobene或meckel指数来测定。为快速筛选有助于减轻亚锡引起的牙齿变色的技术,使用体外实验法,该方法提供对氟化亚锡制剂牙齿变色的预防潜力的定量评估。该方法称为iptsm(体外薄膜茶锈模型),已显示与临床观察结果相关性良好。
体外薄膜茶锈模型(iptsm)是一项技术,其中体外牙斑生物质在三天时间内自积聚的人体受激唾液生长在玻璃棒上。用试剂处理牙斑生物质,以测定各种试剂的潜在牙齿变色程度。此技术的目的在于提供简单而快速的方法来确定化合物是否对牙斑色斑量具有直接功效。此方法采用自积聚的人体唾液生长在磨光玻璃棒上的牙斑,处理5分钟持续时间,接着用茶处理10分钟。该体外模型的试验可在五天内完成,在此期间可评估最多12次处理,包括对照处理。
1.将玻璃棒糙化
依次用240、320、400和600号粒度的碳化硅纸材,在车床上将新玻璃棒(5mm×90mm)自距无尖削端约25mm抛光。初始抛光后,仅在每次测试前,用600号粒度纸材将所述棒抛光。
2.唾液收集和制备
通过石蜡刺激,每日收集得自5-10人小组的唾液,并且在4℃下冷藏,直至需要。小心的集中唾液(不倒入蜡/粘液)并且充分混合。
3.第1天:通过用稀释的hcl酸超声处理,清洁玻璃棒,清洗、干燥,并且用600号粒度碳化硅纸材抛光。再次用di水清洗棒,并且干燥。将棒插入到夹持器中,用处理架上的深度计调节深度,并且用橡胶o形环将棒固定。
在午后,用移液管将已加入0.1重量%蔗糖的7ml唾液移入到浸渍挂架上的16×75mm试管中。仅在第一天将蔗糖加入到唾液中。将棒夹持器置于改进的37℃培养箱中,设计所述培养箱以使糙化的玻璃棒以1rpm速率向试管中浸入1.5cm深度。使棒浸泡过夜。培养箱的设计完整示于附件1中。准备用于第2天的上述牙斑生长介质和高压釜(第2天使用前加入唾液)。
4.第2天:早上将唾液加入到牙斑生长介质中并且充分混合。用移液管将7ml牙斑生长介质移入到新浸渍挂架内的新16/75mm试管中。取出具有已用试管的旧挂架,将新的浸渍挂架放入到培养箱中,并且将棒浸渍最少六小时,之后将棒换入到新鲜唾液中,浸渍过夜。
5.第3天:在第三天早上,用移液管将10mldi水移入到处理挂架第二和第三行的17×100mm试管中。这仅应用于牙粉处理物。冲洗溶液可在处理挂架上配备或可不配备冲水管。用移液管将新鲜的积聚唾液移入到浸渍挂架中并且放置一旁。通过将550ml加入到玻璃烧杯中并且将其在微波炉10分钟,开始制备茶。十分钟结束时,小心地将烧杯从微波炉中取出,并且放入磁力搅拌棒,以驱散可能存在的过热水芯。将5个lipton茶袋和摄氏温度计放入水中,并且在热板上搅拌。该溶液需要监测以确保开始茶处理时,它不高于50℃。在加热并且混合茶处理物的同时,配制牙粉混悬液(1份牙粉对3份水,还称为四分之一稀释)使用手持式匀化器匀化30秒。以10000rpm速率将混悬液离心15分钟。简单处理冲洗液或活性物质溶液。用移液管将7ml50℃茶溶液移入到单独的浸渍挂架上。将5ml上清液/冲洗液加入到处理挂架第一行的16×75mm玻璃试管中。关闭培养箱浸渍机,并且去除旧唾液浸渍挂架。从培养箱中取出所有棒夹持器,并且将棒浸没放入到旧唾液浸渍挂架中以防止变干。一次使用一个棒夹持器,通过在处理挂架中浸泡5分钟来处理。如果适用,通过在处理挂架上包含di水的试管中浸渍2×10秒来洗涤棒。将棒夹持器置于制备好的茶溶液浸渍挂架上并浸泡10分钟。对所有四个棒夹持器重复该过程,将夹持器返回浸渍挂架以防止变干。将新鲜唾液浸渍挂架放入到培养箱中。在处理/茶浸泡后将棒放回至培养箱中,并且在新鲜唾液中浸渍最少1小时。用新鲜处理物/茶/唾液溶液将该处理循环再重复两次,一天共处理3次。最后处理之后,将棒放回至培养箱,并且在新鲜唾液中浸渍过夜。
6.第4天:在第四天早上,关闭培养箱浸渍机构,并且将棒从唾液中取出。使棒干燥,然后称重至最接近0.1mg。记录重量,并且计算平均干燥牙斑生物质重量和标准偏差。将棒放入到包含3ml0.5mkoh的干净无菌可盖盖的试管中,将盖盖紧,并且在37℃下消解过夜。
7.第5天:在第五天,将棒从培养箱中取出,并且使其冷却。将玻璃棒涡旋,以确保所有沉积物匀化。将棒从试管中取出,使溶液过滤通过0.45μm乙酸纤维素注射器过滤器,并且在分光光度计上读取每个棒在380nm处的吸光度值。记录结果并且根据以下公式使用吸光度值计算每次处理的平均吸光度值、每次处理的标准偏差、每mg牙斑的平均吸光度、每mg牙斑的平均吸光度标准偏差,以及相对于对照物的每mg牙斑吸光度增加%,
潜在污渍%=((测试产品abs/生物质-非亚锡对照物abs/生物质)/(高亚锡对照物abs/生物质-非亚锡对照物abs/生物质))*100
实施例1-乙烯基膦酰基-单磷酸盐(vpp)或[乙烯基膦酸磷酸酐]的合成
在氮气下向磁力搅拌的500ml干燥单颈圆底烧瓶中装入乙烯基膦酸(vpa,25.0g,231.5mmol)和300mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。加入三丁胺(64.3g,82.7ml,1.5当量)并在室温下搅拌30分钟,得到浑浊溶液,静置时该浑浊溶液分离成小的上层和大的下层。
在氮气下向配有加料漏斗的第二磁力搅拌的1000ml干燥单颈圆底烧瓶中装入1,1'-羰基二咪唑(cdi)(45.1g,1.2当量),然后装入300mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。接下来,在约两小时内经由加料漏斗将三丁胺/乙烯基膦酸溶液加入到cdi溶液中,并且将所得混合物在室温下搅拌过夜,得到浅黄色均匀溶液。
在氮气下向配有加料漏斗的第三磁力搅拌的2000ml三颈圆底烧瓶中装入h3po4(56.7g,2.5当量),然后装入400mldmf。将所得混合物在室温下搅拌15分钟,得到均匀溶液。向该混合物中加入三丁胺(128.7g,165.4ml,3.0当量)并将所得物搅拌30分钟,得到浑浊溶液。在约2小时内经由加料漏斗向该浑浊溶液中加入来自第二烧瓶的溶液。将所得物在室温下搅拌过夜,得到浅黄色浑浊溶液。在真空(13托)下除去该溶液中的溶剂直至约70℃的最终温度,得到226g浅黄色浆料。
将所得物溶于450ml水中,并用50%naoh(约110g)将ph调节至10.5,得到2相体系。将下层水相与上层有机相分离。除去水相中的水至约70℃的最终温度和13托的真空,得到212g黄色油状物。将该油状物加热至约60℃,并在5分钟内加入300mlmeoh,得到白色沉淀。从沉淀中滗出meoh,然后将沉淀在烘箱中干燥至150.6g。对沉淀的p-nmr显示具有1.22摩尔当量的正磷酸根、0.29当量的焦磷酸根和0.05摩尔当量的起始乙烯基膦酸的预期膦酰基-单磷酸盐产物。h-nmr还显示产物、原料、残余溶剂和约0.4摩尔当量的咪唑。
为了进一步纯化,将沉淀溶于300ml水中。在快速搅拌下,在30分钟内加入400mlmeoh。通过过滤收集所得的白色沉淀,用100mlmeoh冲洗并干燥过夜,得到102.4g。对该沉淀的p-nmr结果显示,其主要为具有0.06摩尔当量的膦酰基-单磷酸根产物和0.29摩尔当量的焦磷酸根的正磷酸盐。
除去水meoh滤液中的溶剂直至约70℃的最终温度和13托的压力,得到81.94g白色固体。该固体显示主要为具有0.077摩尔当量的乙烯基膦酸根和0.091摩尔当量的正磷酸根的乙烯基膦酰基-单磷酸盐。通过以下方式从该白色固体中萃取残余咪唑:在40℃下在300mlmeoh中快速搅拌固体1小时,在溶液热时滤出不溶性固体,然后用50ml室温meoh冲洗所得固体两次,并将所得固体在室温下于高真空下干燥过夜,得到54.8g白色粉末。
对该最终样品的p-nmr显示具有0.05摩尔当量的正磷酸根、0.04当量的焦磷酸根和0.02当量的乙烯基膦酸根的乙烯基膦酰基-单磷酸盐。h-nmr与具有0.02摩尔当量的咪唑和0.09当量的甲醇的乙烯基膦酰基-单磷酸盐产物一致。使用内标,计算出总活性物质为80.8%,其代表82%的收率。
实施例2-甲基-乙烯基膦酰基-单磷酸盐(mvpp)或[甲基乙烯基膦酸磷酸酐]的合成
按照实施例1的过程,以实施例1的1/14摩尔放大比例,用甲基-乙烯基膦酸替代乙烯基膦酸。最终纯度为71.9%,收率为31.2%。
实施例3-(亚甲基膦酰基-单磷酸)-甲基丙烯酸酯[或((甲基丙烯酰氧基)甲基)膦酸磷酸酐]的合成
在氮气下向磁力搅拌的50ml干燥单颈圆底烧瓶中装入(亚甲基膦酸)-甲基丙烯酸酯(0.5g,2.77mmol)和10mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。加入三丁胺(0.77g,1.0ml,1.5当量)并在室温下搅拌30分钟,得到均匀溶液。
在氮气下向第二磁力搅拌的10ml干燥单颈圆底烧瓶中装入1,1'-羰基二咪唑(cdi)(0.54g,1.2当量),然后装入10mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。接下来,在1分钟内将三丁胺/(亚甲基膦酸)-甲基丙烯酸酯溶液加入到cdi溶液中,并将所得混合物在室温下搅拌4小时,得到浅黄色均匀溶液。
在氮气下向第三磁力搅拌的50ml单颈圆底烧瓶中装入h3po4(0.68g,2.5当量),然后装入15mldmf。将所得混合物在室温下搅拌15分钟,得到均匀溶液。向该混合物中加入三丁胺(1.54g,2.0ml,3.0当量)并将所得物搅拌30分钟,得到浑浊溶液。在1分钟内向该浑浊溶液中加入来自第二烧瓶的溶液。将所得物在室温下搅拌过夜,得到浅黄色浑浊溶液。在真空(13托)下除去该溶液中的溶剂直至约65℃的最终温度,得到24.5g浅黄色浆料。
将所得物溶于100ml水中,并且用1nnaoh(约14g)将ph调节至8,得到乳白色体系,随后将其在65℃和13托下浓缩成24.5g浅黄色浆料。在5分钟内将该浆料加入到50mlmeoh中,得到白色沉淀。滗出meoh以除去沉淀,然后在真空下除去meoh,得到7.4g凝胶状固体。对该凝胶状固体的p-nmr显示预期的膦酰基-单磷酸盐产物,其中1当量产物具有0.55摩尔当量的正磷酸根、0.40当量起始膦酸酯的酸酐。
将大部分凝胶状固体在室温下于50mletoh中搅拌1小时,得到不溶性沉淀。将沉淀过滤,用10ml新鲜etoh冲洗两次,并用o/n罩式干燥至238mg固体。对固体的p-nmr显示比率为100:70:10的产物峰双峰(1.1/1.0ppm和-8.4/-8.5ppm)、磷酸盐(2.04ppm)、产物酸酐(2.9ppm),以及其他少量未知物。h-nmr显示干燥的固体与含有约12摩尔%咪唑、残余etoh和其他少量未知物的产物一致。
实施例4-(乙基膦酰基-单磷酸)(丁基)丙烯酰胺或[(2-(n-丁基丙烯酰胺基)乙基)膦酸磷酸酐]的合成
向磁力搅拌的25ml干燥双颈圆底烧瓶中装入正丁胺(6.3ml,63mmol)并在干燥氮气下加热至78℃。加入二乙基乙烯基膦酸盐(1.0ml,6.3mmol)并搅拌过夜。在约45℃和20毫巴下旋转蒸发所得混合物,以通过p-nmr回收1.38g高纯度二乙基乙基膦酸丁胺的物质(92%回收率)。
向磁力搅拌的25ml干燥双颈圆底烧瓶中装入二乙基乙基膦酸丁胺(1.1g,4.6mmol)、2ml二氯甲烷和6ml1nnaoh。将所得物搅拌并在冰浴中冷却。在30分钟内向该烧瓶中滴加2ml二氯甲烷和0.368g丙烯酰氯的混合物。将所得物用10ml二氯甲烷稀释,在分液漏斗中用2×25ml1nhcl、1×25ml饱和nacl萃取,用10ml二氯甲烷冲洗水相。将所得合并的有机相经无水硫酸钠干燥并过滤。在约35℃下通过旋转蒸发除去溶剂,得到0.84g(72%)产物。
通过在冰浴中,在磁力搅拌的100ml双颈烧瓶中,在干燥氮气下将整个批料溶于4ml二氯甲烷中,然后在20分钟内加入1ml二氯甲烷和2ml三甲基溴代硅烷的混合物,以除去该产物上的乙酯基团。然后加入另外的1ml二氯甲烷,然后加入1ml二氯甲烷和1ml三甲基溴代硅烷的混合物。2小时后,加入30mlmeoh并搅拌10分钟,然后加入0.21mg丁基化羟基甲苯的1ml二氯甲烷溶液。在约40℃下通过旋转蒸发除去挥发物。通过溶于50ml二氯甲烷中并用25ml0.1nnaoh和25ml1nnaoh的混合物萃取来纯化所得物。将水相用25ml二氯甲烷第二次萃取,然后用1nhcl酸化至ph1,然后旋转蒸发至接近干燥。将所得残余物用50mletoh稀释并旋转蒸发至接近干燥3次以除去水。然后将所得残余物用10ml戊烷稀释并蒸发至接近干燥2次以除去残余etoh。最终回收率接近定量。
如实施例3中那样进行膦酰基-磷酸根基团的添加。略微修改纯化步骤。对粗反应混合物进行二乙醚(1体积当量)萃取,然后在30℃至35℃下真空除去溶液。将残余物溶于25ml水中,并且用1nnaoh将ph调节至7,然后在40℃至45℃下真空除去水,以留下液体残余物。接下来,将100ml甲醇加入到残余物中,从而产生沉淀,收集沉淀并真空干燥,得到大约9.1g,通过p-nmr测定为80%活性物质。
实施例5-(4-乙烯基苄基)膦酰基-单磷酸盐或[(4-乙烯基苄基)膦酸磷酸酐]的合成
在氮气下向磁力搅拌的50ml干燥单颈圆底烧瓶中装入(4-乙烯基苄基)膦酸(4.0g,20.2mmol)和20mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。加入三丁胺(5.6g,7.2ml,1.5当量)并在室温下搅拌30分钟,得到均匀溶液。
在氮气下向第二磁力搅拌的10ml干燥单颈圆底烧瓶中装入1,1'-羰基二咪唑(cdi)(4.9g,1.2当量),然后装入25mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。接下来,在1分钟内将三丁胺/(4-乙烯基苄基)膦酸溶液加入到cdi溶液中,并将所得混合物在室温下搅拌4小时,得到浅黄色均匀溶液。
在氮气下向第三磁力搅拌的100ml单颈圆底烧瓶中装入h3po4(5.94g,3.0当量),然后装入25mldmf。将所得混合物在室温下搅拌15分钟,得到均匀溶液。向该混合物中加入三丁胺(13.1g,16.8ml,3.5当量)并将所得物搅拌30分钟,得到浑浊溶液。在1分钟内向该浑浊溶液中加入来自第二烧瓶的溶液。将所得物在室温下搅拌过夜,得到浅黄色溶液。在真空(13托)下除去该溶液中的溶剂直至约65℃的最终温度,得到49.8g浅黄色浆料。
将所得物加入到30ml水中,并且用1nnaoh(约127g)将ph调节至8.5,得到乳白色体系,随后将其在65℃和13托下浓缩成58.2g浅黄色浆料。在20分钟内将该浆料加入到40mlmeoh中,得到白色沉淀。对该糊状物的p-nmr显示其为约95%的磷酸盐。滗出meoh以除去沉淀,然后除去meoh,得到7.23g白色糊状物。对该白色糊状物的pnmr显示预期的膦酰基-单磷酸盐产物,其中1当量产物具有0.16摩尔当量的正磷酸根、0.05当量的焦磷酸根、0.25当量的起始膦酸酯的酸酐和0.065当量的起始膦酸酯。
将大部分白色糊状物在室温下于75mlmeoh中搅拌1小时。糊状物的一部分溶解,而一部分保持不溶解。将不溶解的部分过滤并用10ml新鲜meoh冲洗两次。将所得固体在高真空o/n下干燥,得到1.97g固体。对固体的p-nmr显示比率为100:70:10的产物峰双峰(1.1/1.0ppm和-8.4/-8.5ppm)、磷酸盐(2.04ppm)、产物酸酐(2.9ppm),以及其他少量未知物。h-nmr显示干燥的固体与含有约12摩尔%咪唑、残余etoh和其他少量未知物的产物一致。最终固体的收率为理论值的23.7%。
实施例6-(双(亚甲基膦酸酯酸酐)氨基丙基)-甲基丙烯酸酯聚合物或[4-(3-(甲基丙烯酰氧基)丙基)-1,4,2,6-氧氮二膦-2,6-双(醇盐)2,6-二氧化物聚合物]的合成
在氮气下向磁力搅拌的250ml干燥三颈圆底烧瓶中装入(双(亚甲基膦酸)氨基丙基)-甲基丙烯酸酯(3.0g,9.06mmol)和100mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。加入三丁胺(5.03g,6.5ml,3.0当量)并在室温下搅拌30分钟,得到均匀溶液。
在氮气下向配有加料漏斗的第二磁力搅拌的100ml干燥单颈圆底烧瓶中装入1,1'-羰基二咪唑(cdi)(2.2g,1.5当量),然后装入40mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。在大约一小时内经由加料漏斗将cdi溶液加入到第一烧瓶中,并且将所得混合物在室温下搅拌过夜,然后静置1周,得到白色沉淀。通过过滤收集沉淀,在100ml水中浆化,用1nnaoh将ph调节至约等于9,得到混浊溶液。将该溶液在流动空气下蒸发过夜至2.4g。h-nmr和p-nmr的结果显示沉淀为具有一些单体的聚合物。p-nmr显示比率为36:53:11的聚合物目标酸酐(12-13ppm处)和起始二酸(6-7ppm处)以及单体峰(11.8-12ppm处)。将大部分沉淀在100ml水中超声处理1小时,得到浑浊溶液,使用具有0.22μmpvdf滤盘的250mlstericupdurapore过滤该浑浊溶液,得到澄清溶液。将其补充到250ml,并通过在thermoscientificslide-a-lyzer透析烧瓶(2kmwco,250ml)中用5加仑ro水(用1nnaoh将ph调节至8.5)透析7天来纯化,冷冻干燥后得到1.29g白色固体(17-df-5835-5)。p-nmr显示在12-13ppm处具有宽酸酐峰并且在6.4-7.4ppm处具有二酸峰,摩尔比为39.2:60.8。活性计算为87.8%的聚合物和12.2%的水/无活性物质。
实施例7-(乙基膦酰基-单磷酸)-甲基丙烯酸酯或[(2-(甲基丙烯酰氧基)乙基)膦酸磷酸酐]的合成
在氮气下向磁力搅拌的100ml干燥单颈圆底烧瓶中装入(乙基膦酸)-甲基丙烯酸酯(3g,15.5mmol)和30mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。加入三丁胺(4.3g,5.5ml,1.5当量)并在室温下搅拌30分钟,得到均匀溶液。
在氮气下向第二磁力搅拌的25ml干燥单颈圆底烧瓶中装入1,1'-羰基二咪唑(cdi)(3.76g,1.5当量),然后装入20mldmf。将所得混合物在室温下搅拌10分钟,得到均匀溶液。接下来,将三丁胺/(乙基膦酸)-甲基丙烯酸酯溶液加入到cdi溶液中,并将所得混合物在室温下搅拌4小时,得到浅黄色均匀溶液。
在氮气下向第三磁力搅拌的500ml单颈圆底烧瓶中装入h3po4(4.55g,3.0当量),然后装入25mldmf。将所得混合物在室温下搅拌15分钟,得到均匀溶液。向该混合物中加入三丁胺(12g,15.4ml,4.2当量)并将所得混合物搅拌30分钟,得到浑浊溶液。在1分钟内向该浑浊溶液中加入来自第二烧瓶的溶液。在搅拌约1小时时,开始形成白色沉淀。将所得物在室温下搅拌过夜,形成另外的沉淀。p-nmr显示比率为100:427:12:15的产物峰双峰(7.2/7.3ppm和-8.75/-8.84ppm)、膦酸盐(2.14ppm)、产物酸酐(8.9ppm)和焦磷酸盐(-9.26ppm)。
在搅拌下,在30分钟内向粗制rx溶液(约等于90.7g)中加入200ml乙醚,得到白色沉淀,将其通过过滤收集,用另外的醚冲洗并在室温下真空(<1托)干燥过夜,得到7.85g白色沉淀。在搅拌下,在30分钟内向所得滤液中加入另外的200ml乙醚,得到具有自由流动顶层和下部粘稠油状物层的两层体系。滗出顶层,并且将下部油层在室温下真空(<1托)干燥过夜,得到2.33g蜡质固体。在搅拌下,在30分钟内向滗析层中加入另外的400ml醚,得到浑浊溶液。将浑浊溶液置于冷冻机(-15℃)中过夜,得到澄清的自由流动顶层和粘稠油状物下层。滗出顶层,并且将下部油层在真空(<1托)和室温下干燥过夜1小时,得到1.19g蜡质固体。
通过h-nmr显示白色沉淀为摩尔比为100:1150:220的产物:咪唑:三丁胺的混合物,而p-nmr显示摩尔比为100:625:35的产物:磷酸盐:焦磷酸盐的混合物。
h-nmr显示第一蜡质固体为摩尔比为100:230:170的产物:咪唑:三丁胺的混合物。p-nmr显示摩尔比为100:89的产物:磷酸盐的混合物。
h-nmr显示第二蜡质固体为摩尔比为100:100:150的产物:咪唑:三丁胺的混合物。p-nmr显示摩尔比为100:79的产物:磷酸盐的混合物。
将蜡质固体合并并溶于50ml去离子水中。用19.3g1nnaoh将所得溶液的ph从2.9调节至8.6,得到浑浊溶液。将该溶液用50ml乙醚萃取1次。用另外的1nnaoh将所得水层的ph=7.5修整至8.0。在室温和20托下,在旋转蒸发器上从水层中除去残余的醚。通过冷冻干燥从水层中除去水,得到2.61g棕褐色固体。
h-nmr和p-nmr的结果显示摩尔比为100:160:50的产物:咪唑:nbut3的混合物。p-nmr显示摩尔比为100:101的产物:磷酸盐的混合物。
将棕褐色固体在50mlmeoh中搅拌30分钟,得到不溶性固体。通过过滤收集固体,用2×10ml新鲜meoh冲洗,并在室温下于<1托下干燥过夜,得到1.79g乳白色固体。h-nmr的结果与含约1摩尔%咪唑的产物一致。lcms显示出在273处与m+h质子化形式一致的质量。甲醇萃取物的h-nmr显示其主要为含约3摩尔%产物的咪唑。
通过结合h-nmr和p-nmr计算活性,并发现其为74.4%。
实施例8-(丙基膦酰基-单磷酸)-甲基丙烯酸酯或[(3-(甲基丙烯酰氧基)丙基)膦酸磷酸酐]的合成
按照实施例7的过程,用5.5g(26.4mmol)(丙基膦酸)-甲基丙烯酸酯代替(乙基膦酸)-甲基丙烯酸酯。所有试剂按比例缩放以保持摩尔当量相同。在最终蒸发后,收集5.17g乳状固体,并且显示活性为67.8%。
实施例9-(乙基膦酰基-单磷酸)-丙烯酰胺或[(2-丙烯酰胺基乙基)膦酸磷酸酐]的合成
按照实施例5的过程,用(丙烯酰氨基)乙基膦酸(37mmol)代替乙烯基苄基膦酸盐来形成粗制黄色溶液,所有试剂的量以与实施例5同等的摩尔比增加。
粗制溶液的纯化过程由实施例5修改而来。在室温下,用流动的干燥氮气部分除去dmf,得到46.8g粘稠黄色油状物。将该油状物溶于约75ml水中,并通过在20分钟内加入1nnaoh将ph调节至8。形成9.4g三丁胺的小有机层并滗出。在流动的干燥氮气下将水相进一步干燥至147.5g,然后在30分钟内加入220mlmeoh,得到白色沉淀。将沉淀过滤,并且将所得滤液干燥,得到22.7g棕色糊状物,其pnmr显示大部分为产物。将棕色糊状物在室温下于100mletoh中在剧烈搅拌下浆化6小时。形成固体并通过过滤收集,用新鲜etoh2×25ml冲洗,并在<1托下干燥过夜,得到10.94g棕褐色固体。nmr表明固体含有69%膦酰基-单磷酸盐产物。
实施例10-(亚甲基膦酰基-单磷酸)-丙烯酸酯或[((丙烯酰氧基)甲基)膦酸磷酸酐]的合成
按照实施例3的过程,用(亚甲基膦酸)-丙烯酸酯代替(亚甲基膦酸)-甲基丙烯酸酯
实施例11-(乙基膦酰基-单磷酸)-乙烯基醚或[(2-(乙烯氧基)乙基)膦酸磷酸酐]的合成
向干燥、隔膜密封、氮气吹扫、磁力搅拌的250ml三颈圆底烧瓶中装入二乙基(2-(乙烯氧基)乙基)膦酸盐(3g,14.4mmol)和30mlch2cl2,并冷却至0-5℃。在1分钟内向该烧瓶中加入溴代三甲基硅烷(5.7ml,43.2mmol,3.0当量)。加入后,将溶液在室温下搅拌2小时,并在30℃和<1托下除去溶液中的溶剂,得到4.59g黄色油状物。向其中加入15g三乙胺、30gmeoh和60mg已在干冰和丙酮上预冷却的吩噻嗪(抑制剂)。在持续搅拌下使所得物升温至室温,然后在室温下真空处理1小时以除去溶剂和挥发物,得到3.84g粘稠浑浊黄色油状物。p-nmr和h-nmr与胺单三乙胺盐一致。
按照实施例7的过程,产生并纯化(乙基膦酰基-单磷酸)-乙烯基醚,在甲醇萃取后得到4.04g棕褐色固体。h-nmr的结果与含约5摩尔%咪唑的产物一致。p-nmr与100:112的产物膦酰基-单磷酸盐与残余磷酸盐的比率一致。lcms显示出在231处与m+h质子化形式一致的质量。通过结合h-nmr和p-nmr计算活性,并发现其为52%。
实施例12-(乙基膦酰基-单磷酸)-丙烯酸酯或[(2-(丙烯酰氧基)乙基)膦酸磷酸酐]的合成
向干燥磁力搅拌的1l三颈圆底烧瓶中装入(2-羟乙基)膦酸二甲酯(24.7g,16mmol)、三乙胺(17.8g,176mmol)和400mlch2cl2,并冷却至0-5℃。在1.5小时内,同时保持反应温度为0-5℃,向该烧瓶中加入丙烯酰氯(14.95g,16.51mmol)的100mlch2cl2溶液。加入完成后,将反应温度在0-5℃下再保持2小时,然后升温至室温并搅拌过夜。
将所得的浅棕色浑浊溶液用2×200ml去离子水萃取,将油层在无水mgso4上干燥,然后过滤。除去滤液中的溶剂,得到30.5g棕色油状物。h-nmr、c-nmr和p-nmr的结果与第一中间体(乙基,二甲基膦酸)-丙烯酸酯一致。收率为91.4%。
将上述棕色油状物装入具有250ml二氯甲烷的干燥磁力搅拌的500ml三颈圆底烧瓶中。将烧瓶和内容物冷却至10℃,并在30分钟内加入67.3g(3当量)溴代三甲基硅烷。使烧瓶升温至室温并搅拌过夜。在30℃下除去所得溶液中的溶剂,然后在高真空(<1托)下搅拌过夜,得到37g轻质油状物。在室温下,在10分钟内向该油状物中加入200ml甲醇,然后在室温下搅拌3小时。在30℃下除去所得溶液中的溶剂,然后在高真空(<1托)下搅拌过夜,得到26.1g粘稠棕褐色油状物。hnmr和pnmr与产物一致。收率为98.9%。
按照实施例5的过程,用(丙烯酰氧基)乙基膦酸代替乙烯基苄基膦酸盐来形成粗制黄色溶液。pnmr和hnmr显示期望产物的收率为72%。
实施例13-混合的乙烯基膦酰基-磷酸盐的合成
按照实施例1的过程,以实施例1的1/12摩尔放大比例,用焦磷酸替代磷酸。在除去dmf溶剂以得到黄色油状物并加入1nnaoh后,用氮气鼓泡过夜后收集24.1g白色固体。pnmr表明该样品含有乙烯基膦酰基-焦磷酸盐(vppp)、乙烯基膦酰基-单磷酸盐、原料、原料酸酐、磷酸盐、焦磷酸盐和三磷酸盐。乙烯基膦酰基焦磷酸盐:乙烯基膦酰基-单磷酸盐的比率为1:1.7。
实施例14乙烯基磺酸甲酯(vsme)的合成
在氮气下向配备有加料漏斗和温度计的磁力搅拌的500ml干燥三颈圆底烧瓶中装入250ml甲醇,并且冷却至0℃。在15分钟内将2-氯乙烷磺酰氯(aldrich)加入烧瓶中,未观察到放热。接下来,在2小时内以一定速率加入25%naome/meoh(aldrich)以保持约0℃的温度。在加入过程中形成白色沉淀(nacl)。将所得物在0℃下再搅拌一小时,然后使其升温至室温并搅拌过夜。通过过滤除去沉淀,并除去滤液中的溶剂,得到20.33g白色凝胶。将该凝胶在200mlch2cl2中浆化1小时。将所得物过滤并除去滤液中的溶剂,得到9.47g棕褐色油状物。
1h-nmr和13c-nmr显示3:0.6比率的期望产物vsms和2-甲氧基乙烷-1-磺酸甲酯的混合物,产物占79.8重量%。h-nmr还显示在约10ppm处的酸峰。通过石蕊在1ml水中测试0.05g棕褐色油状物,显示ph为约1。
将8.8g棕褐色油状物溶于100mlch2cl2中,并在5g碳酸氢钠中搅拌。将所得物过滤并除去滤液中的溶剂,得到8.02g浅黄色澄清油状物。
通过石蕊在1ml水中测试0.05g黄色澄清油状物,显示ph约为6-7。vsme与2-甲氧基乙烷-1-1磺酸甲酯的比率相同,得到79.8%活性物质。
实施例15乙烯基苄基磺酸钠的合成
向配备有加热套、加料漏斗和回流冷凝器的磁力搅拌的250ml干燥三颈圆底烧瓶中装入9.5g亚硫酸钠(75.5mmol)和100ml水。在氮气下将所得溶液加热至100℃。接下来,在30分钟内加入4-乙烯基苄基氯(9.6g,62.9mmol)的15ml丙酮溶液。使所得物回流12小时,冷却至室温,并且使其静置过夜,没有产生沉淀。接下来,在快速搅拌下,加入100ml丙酮,得到糊状下层。滗出水/丙酮上清液。用25ml新鲜丙酮冲洗该糊状物,然后将其滗出。将该糊状物在真空、14托和室温下干燥过夜,得到8.5g固体。通过1h-nmr显示该干燥层主要为均聚物。将水/丙酮滗析层蒸发至约75ml,得到白色沉淀。通过过滤收集沉淀,用2×25ml丙酮冲洗,并在真空、14托和室温下干燥过夜,得到2.14g固体。
该第二沉淀的1h-nmr与接近100%活性的单体产物一致。
实施例16乙烯基膦酸(vpa)和乙烯基磺酸钠(svs)的共聚
在圆底烧瓶中装入vpa(2.0g,18.5mmol)和svs(25%水溶液,7.9g,15.2mmol),svs与vpa的初始摩尔比为45至55。用氮气将烧瓶吹扫15分钟,并且加热至90℃。另外制备包含2,2'-偶氮双(2-甲基丙脒)二盐酸盐(aaph,aldrich,在1.2ml水中25.8mg,相对于所加入的总单体为0.3摩尔%)和1-辛硫醇(cta,aldrich,在1.2ml水中55.6mg,相对于所加入的总单体为1.1摩尔%)的两种独立的水溶液。然后在6小时期间内,每30分钟将这两种溶液加入到包含单体的热搅拌烧瓶中。最后一次加入后,使所得溶液在90℃下搅拌过夜。
对粗反应溶液运行1h-nmr和31p-nmr。观察到95%至99%的典型单体转化率,膦酸根基团在约31ppm处具有宽p聚合物峰。
将粗反应溶液在水中稀释至1重量%的聚合物,并将ph调节至6。将这些溶液用2k分子量截留的渗析膜对反渗透水透析5天至7天。
在真空下除去所得溶液中的水,得到白色至乳白色固体,将其在真空烘箱中进一步干燥过夜,得到2.74g固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。基于此分析,聚合物包含得自svs的55.7摩尔%的重复单元,以及得自vpa的44.3摩尔%的重复单元。水含量计算为9.6重量%。渗析后聚合物中单体的总回收率计算为57摩尔%。
实施例17乙烯基膦酸与乙烯基磺酸钠(svs)的共聚
对于不同起始比率的vsa和vpa,重复实施例16的过程。不同起始比率和总收率所得的聚合物组合物(包括实施例16)如下表1所示。使用聚合物标准服务(pss)mcx1000a柱以及wyattheleosii光散射检测器和wyattoptilab示差折光率检测器两者的wyatt凝胶渗透色谱(gpc)系统,用于使用内部wyattastra6软件计算聚合物分子量。
表1
实施例18乙烯基膦酸(vpa)和丙烯酸(aa)的共聚
使用19mmolvpa初始进料的1.5ml水溶液重复实施例x中的过程。每30分钟加入溶于1.6ml水中的28.5mmol丙烯酸(0.3ml)以及0.1mlaaph和cta。在加入的中途加入另外3ml水。透析后收集的最终聚合物为2.87g,并且发现其为30%的膦酸盐和70%的丙烯酸酯。
实施例19乙烯基膦酰基-单磷酸盐(vpp)和乙烯基磺酸钠(svs)的共聚
将vpp(实施例1,2.05g活性物质,8.87mmol)和svs(25%水溶液,3.77g,7.25mmol)装入圆底烧瓶中,其中svs与vpp的初始摩尔比为45至55,并用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃,此时将过硫酸铵(aps,aldrich,183mg,相对于总单体为5%)加入0.50ml水中。将所得物在60℃下搅拌24小时。
对粗反应溶液运行1h-nmr和31p-nmr。观察到典型单体转化率为95%至99%,膦酸根基团在约18ppm至23ppm处具有宽p聚合物峰,并且与膦酸根基团键合的磷酸根在-6ppm至-10ppm处具有宽p聚合物峰。
将粗反应溶液在水中稀释至1重量%的聚合物,并将ph调节至8.5。将这些溶液用2k分子量截留的渗析膜对反渗透水透析5天至7天。
通过冷冻干燥从产物中除去水,得到2.22g白色固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h-nmr和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。p-nmr显示在约18ppm至23ppm和在-6ppm至-10ppm处具有约1:1比率的宽膦酰基-磷酸根峰并且在约26-28ppm处具有膦酸根峰。基于18-23ppm和26-28ppm处的31p-nmr面积,膦酰基-磷酸根:膦酸根比率为94.9:5.1。基于此分析,聚合物包含得自svs的56摩尔%的重复单元、得自vpp的42摩尔%的重复单元和得自vpa的2摩尔%重复单元。水含量计算为23重量%。透析后聚合物中单体的总回收率计算为65摩尔%。
实施例20乙烯基膦酰基-单磷酸盐(vpp)和乙烯基磺酸钠(svs)的共聚
对于不同起始比率的vsa和vpp,重复实施例19的过程。不同起始比率和总收率所得的聚合物组合物(包括实施例19)如下表2所示。
表2
实施例21甲基-乙烯基膦酰基-单磷酸盐(mvpp)和乙烯基磺酸钠(svs)的共聚
使用mvpp(实施例2)代替vpp,并且mvs与mvpp的比率分别为55至45,重复实施例19的过程,但有以下变化。
在24小时的运行时间,经由nmr的mvpp单体转化率约为75%,因此另外加入3摩尔%的aps水溶液,并将反应物在60℃下另外搅拌24小时。此时,mvpp单体转化率为约95%。
如实施例19所述进行透析和冷冻干燥。
基于此nmr分析,聚合物包含得自svs的62摩尔%的重复单元、得自mvpp的35摩尔%的重复单元和得自甲基乙烯基膦酸的3摩尔%的重复单元。水含量计算为10.3重量%。透析后聚合物中单体的总回收率计算为65摩尔%。
实施例22乙烯基膦酰基-单磷酸盐的均聚
将6mlvpp(实施例1,16.4mmol)的水溶液和碳酸氢钠(0.69g,8.2mmol)装入25ml圆底烧瓶中,然后用氮气吹扫15分钟。将过硫酸铵(aps,186.6mg)溶于0.50ml水中并加入混合物中。使所得溶液在60℃下搅拌6小时。此时,nmr显示25%的聚合单体。加入另外的186.6mg的aps的0.50ml水溶液。使所得物在60℃下搅拌总共24小时。nmr显示没有剩余的单体。
将粗反应溶液用500ml水稀释,得到的ph为8.7。将该溶液用2k分子量截留的渗析膜对调节ph为8.5的反渗透水进行透析。
在真空下除去所得溶液中的水,得到白色至乳白色固体,将其在真空烘箱中进一步干燥过夜,得到2.8g固体。p-nmr显示仅vpp而无vpa。所得物为91重量%的聚合物,剩余的为水和杂质。透析后聚合物中单体的总回收率计算为58摩尔%。
实施例23乙烯基膦酰基-单磷酸盐(vpp)和2-丙烯酰氨基-2-甲基丙烷磺酸钠(amps)的共聚
将vpp(实施例1,6.55mmol)和水2ml装入圆底烧瓶中,并用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃保持15分钟,得到均匀溶液。将过硫酸铵(aps,149.3mg)溶于1.2g水中。在总共6小时内,每30分钟向反应中加入0.1mlaps溶液和0.206mlamps(3g50%的溶液,6.55mmol)。将所得物在60℃下搅拌24小时。
将粗反应溶液用250ml水稀释,并用2k分子量截留的渗析膜对反渗透水透析6天。
通过冷冻干燥从产物中除去水,得到2.66g白色固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h-nmr和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。p-nmr显示在约18ppm至23ppm和在-6ppm至-10ppm处具有约1:1比率的宽膦酰基-磷酸根峰并且在约26-28ppm处具有膦酸根峰。基于18-23ppm和26-28ppm处的31p-nmr面积,膦酰基-磷酸根:膦酸根比率为98:2。基于此分析,聚合物包含得自amps的64.2摩尔%重复单元、得自vpp的35.1摩尔%重复单元和得自vpa的0.7摩尔%重复单元。水含量计算为13.3重量%。
实施例24乙烯基膦酰基-单磷酸盐(vpp)和3-磺基丙基丙烯酸酯钾盐(spa)的共聚
按照实施例23的过程,用spa(aldrich)代替amps。
冷冻干燥产物,得到2.04g白色固体。
基于nmr分析,聚合物包含得自spa的62摩尔%重复单元、得自vpp的36摩尔%重复单元和得自vpa的2摩尔%重复单元。水含量计算为15.5重量%。
实施例25vpp与丙烯酰胺的共聚
将vpp(实施例1,9.9mmol)和水3ml装入圆底烧瓶中,并用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃保持15分钟,得到均匀溶液。将过硫酸铵(aps,225.9mg)溶于1.2g水中。每30分钟将丙烯酰胺(aldrich,9.9mmol)溶于1.5g水中,在总共6小时内向反应中加入0.1mlaps溶液和0.125ml丙烯酰胺溶液。将所得物在60℃下搅拌24小时。通过nmr监测进展。
将粗反应溶液用250ml水稀释,并用2k分子量截留的渗析膜对反渗透水透析5天。
通过冷冻干燥从产物中除去水,得到2.85g白色固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h-nmr和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。p-nmr显示在约18ppm至23ppm和在-6ppm至-10ppm处具有约1:1比率的宽膦酰基-磷酸根峰。在约26-28ppm处未观察到膦酸根峰。基于此分析,聚合物包含得自丙烯酰胺的53摩尔%的重复单元、得自vpp的47摩尔%的重复单元。水含量计算为16重量%。
实施例26vpp与vsms的共聚
将vpp(实施例1,7.67mmol)、碳酸氢盐(aldrich11.5mmol)和5ml水装入圆底烧瓶中,并用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃保持15分钟,得到均匀溶液。将过硫酸铵(aps,174.9mg)溶于1.2g水中。在总共3小时内,每15分钟向反应中加入0.1ml的aps溶液和0.084ml的vsme(乙烯基磺酸甲酯(实施例14,79.8%活性物质,总共7.67mmol))。将所得物在60℃下另外搅拌3小时。将粗反应溶液用250ml水稀释,并用2k分子量截留的渗析膜对ph为8.5的反渗透水透析5天。
通过冷冻干燥从产物中除去水,得到2.05g白色固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h-nmr和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。p-nmr显示在约18ppm至23ppm和在-4ppm至-10ppm处具有约1:1.1比率的宽膦酰基-磷酸根峰但在约26-28ppm处没有膦酸根峰。总磷含量为40.8%。来自msme的甲基质子在1hnmr中是可见的,并且允许定量vsme的水解。基于总分析,聚合物包含得自vsme的39摩尔%的重复单元、得自vsa的20摩尔%和得自vpp的41摩尔%的重复单元。所得物为73重量%的聚合物,剩余的为水和杂质。
实施例27(膦酰基-单磷酸酯乙基)(丁基)丙烯酰胺与amps的共聚
如实施例23那样使(膦酰基-单磷酸酯乙基)(丁基)丙烯酰胺(实施例4,21.6mmol)和amps(23.6mmol)聚合。将粗反应溶液用1k分子量截留的渗析膜对反渗透水透析过夜,然后用0.5mnacl膨胀2小时,然后用0.05mnacl膨胀1小时。冷冻干燥后,收集到11.5克物质。如在其他实施例中的nmr发现,聚合物是得自amps的约67摩尔%的重复单元和得自(膦酰基-单磷酸酯乙基)(丁基)丙烯酰胺的33%的重复单元。固体包含约16重量%的水,并且为55重量%的聚合物。
实施例28vpp与vpa的共聚
将vpa(1.2g,11.1mmol)和6ml水装入25ml圆底烧瓶中。在60分钟内加入碳酸氢钠(2.8g,33.3mmol),然后用氮气吹扫烧瓶,在室温下搅拌过夜。加入vpp(实施例1,11.1mmol),将溶液用氮气吹扫并加热至60℃,得到浑浊溶液。将过硫酸铵(aps,253.5mg)溶于0.75ml水中并加入混合物中。使所得溶液在60℃下搅拌6小时。此时,nmr显示所有单体的40%聚合。加入另外的253.3mg的aps的0.75ml水溶液。使所得物在60℃下搅拌总共24小时。nmr显示剩余10%的单体。
将粗反应溶液用500ml水稀释,得到的ph为8.7。将该溶液用2k分子量截留的渗析膜对调节ph为8.5的反渗透水进行透析。
在真空下除去所得溶液中的水,得到白色至乳白色固体,将其在真空烘箱中进一步干燥过夜,得到2.66g固体。p-nmr显示聚合物中vpp:vpa的比率为61:39。所得物为88重量%的聚合物。
实施例29vpp与丙烯酸甲酯的共聚
将vpp(实施例1,9.9mmol)和水4ml装入圆底烧瓶中,并用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃保持15分钟,得到均匀溶液。将过硫酸铵(aps,225.7mg)溶于1.2g水中。在总共6小时内,每30分钟向反应中加入0.1mlaps溶液和0.073ml丙烯酸甲酯(aldrich,9.9mmol,总共0.88ml)溶液。在3小时时,开始形成乳白色。将所得物在60℃下搅拌24小时,得到乳白色溶液。通过nmr监测进展并显示在24小时时剩余20%的vpp并且没有剩余的丙烯酸甲酯。
将反应溶液加入到20ml另外的水中,然后在5分钟内,在快速搅拌下加入5mlmeoh。使所得物在室温下静置10分钟,得到白色沉淀。将沉淀过滤,并且从滤液中除去meoh。
将滤液用250ml水稀释,并用2k分子量截留的渗析膜对反渗透水在约6.5的ph下透析5天。
通过冷冻干燥从产物中除去水,得到1.4g白色固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。分析表明,所得聚合物为29%的丙烯酸甲酯、16%的丙烯酸酯、50%的vpp和5%的乙烯基膦酸盐。所得固体为77重量%的聚合物。
实施例30(4-乙烯基苄基)膦酰基-单磷酸盐与svs的共聚
将(4-乙烯基苄基)膦酰基-单磷酸盐(vbpp,实施例5,4.35mmol)、碳酸氢盐(aldrich183mg,8.2mmol)和svs(aldrich,25%水溶液,8.1mmol)加入圆底烧瓶中,并且用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃,此时释出气体。增量地加入另外的碳酸氢盐(总共300mg),直到没有观察到另外的排气。将过硫酸铵(aps,aldrich,284mg,相对于全部单体为10%)加入0.5ml水中。将所得物在60℃下搅拌24小时。
对粗反应溶液运行1h-nmr和31p-nmr。聚合物组成为约25/75的svs/vbpp。
将粗反应溶液用500ml水稀释,并用1nnaoh将ph调节至8.5。将该溶液用2k分子量截留的渗析膜对反渗透水透析6天。通过冷冻干燥从产物中除去水,得到1.85g白色固体。
通过在d2o溶液中用纯化的聚合物和磷酸三甲酯(tmp)制备nmr样品来测定聚合物中的膦酸根含量。运行1h和31p-nmr,根据内标(tmp)相对于聚合物峰和水的h峰和p峰,计算膦酸根含量。p-nmr显示在约13ppm至15ppm和在-5ppm至-7ppm处具有约1:1比率的宽膦酰基-磷酸根峰并且在约21-23ppm处具有膦酸根峰。基于13-15ppm和21-23ppm处的31p-nmr面积,膦酰基-磷酸根:膦酸根比率为93:7。基于此分析,聚合物包含得自svs的30摩尔%的重复单元、得自vbpp的65摩尔%的重复单元和得自(4-乙烯基苄基)膦酸盐的5摩尔%的重复单元。水含量计算为18重量%。
实施例31(膦酰基-单磷酸酯乙基)-丙烯酰胺与svs的共聚
将svs(aldrich,25%水溶液,4.73mmol)装入圆底烧瓶中,并且用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃。将过硫酸铵(aps,aldrich,141mg,相对于全部单体为5%)加入1.0g水中。将膦酰基-单磷酸酯(乙基)-丙烯酰胺(实施例9,4.95mmol)加入5.25g水中。在3小时内,每20分钟向烧瓶中加入svs、0.1mlaps溶液和1.0ml(膦酰基-单磷酸酯乙基)-丙烯酰胺。将所得物搅拌并在60℃下另外搅拌4小时。
将粗反应溶液用500ml水稀释,并用1nnaoh将ph调节至8.5。将该溶液用2k分子量截留的渗析膜对反渗透水透析6天。通过冷冻干燥从产物中除去水,得到3.4g棕褐色固体。对粗反应溶液运行h-nmr和p-nmr。聚合物组成为约62:36:2的svs:(膦酰基-单磷酸酯乙基)-丙烯酰胺:(膦酸酯乙基)-丙烯酰胺。
实施例32vpa和aa的共聚物的后聚合改性
通过在配备有加热器和磁力搅拌的250ml单颈圆底烧瓶中在150mlmeoh中回流2.3克,将得自实施例18的聚合物酯化。回流1小时后,加入短程蒸馏头并除去约1/3的meoh。然后将该meoh用新鲜的无水meoh代替,总共4次。该过程产生约47%的丙烯酸重复单元的甲酯转化率。接下来,加入2滴浓硫酸,并将溶液回流48小时。通过hnmr,该过程将总甲酯含量增加至83%。此外,通过pnmr,约9%的膦酸酯被转化为单甲基膦酸酯。
在氮气下向磁力搅拌的50ml干燥单颈圆底烧瓶中装入含甲酯的聚合物(0.5,1.63mmolp)和15mldmf。将所得混合物在室温下搅拌过夜,得到溶胀的聚合物球。接下来,加入三丁胺(0.78ml,相对于p单体为2.0当量)并在室温下搅拌过夜,得到均匀溶液。将cdi(330mg,相对于p单体1.25当量)和5mldmf预混合并加入到溶液中。将所得混合物搅拌过夜,得到均匀溶液。
将h3po4(479mg,3当量)、三丁胺(1.26ml,3.5当量)和5mldmf混合并超声处理,然后加入到含聚合物的溶液中。将所得物在室温下搅拌过夜。在真空(9托)下除去所得溶液中的溶剂直至约60℃的最终温度。
将所得物溶于50ml的1nnaoh中,得到ph13.15的溶液,并搅拌过夜。用流动干燥氮气除去所得物中的水,得到白色糊状物。在1小时内将该糊状物溶于60mlmeoh中,收集所得固体并干燥至2.52克。
将粗制固体溶于水中,将ph调节至9,并且如先前实施例中所述将所得溶液透析。冻干后收集到0.67g白色蓬松固体。pnmr显示膦酰基-单膦酸根基团的收率为初始膦酸根基团的约20%。
实施例33(膦酰基-单磷酸酯乙基)-甲基丙烯酸酯与svs的共聚
按照实施例31的过程,使用5.7mmol的svs和3.79mmol的得自实施例7的(膦酰基-单磷酸酯乙基)-甲基丙烯酸酯进行。冷冻干燥后,收集到1.5g白色固体,聚合物为86%,水/无活性物质为14%。聚合物组成为约63:35:2的svs:(膦酰基-单磷酸酯乙基)-甲基丙烯酸酯:(膦酸酯乙基)-甲基丙烯酸酯。
实施例34(丙基膦酰基-单磷酸)-甲基丙烯酸酯与svs的共聚
按照实施例31的过程,使用3.7mmol的svs和3.5mmol的得自实施例8的(丙基膦酰基-单磷酸)-甲基丙烯酸酯进行。冷冻干燥后,收集到1.84g白色固体,聚合物为86%,水/无活性物质为14%。聚合物组成为约56:44的svs:(丙基膦酰基-单磷酸)-甲基丙烯酸酯。
实施例35vpa均聚物的后聚合改性
将聚(乙烯基膦酸)(500mg)加入到100ml圆底烧瓶中,然后加入甲醇(20ml)。向混合物中加入三丁胺(1.1ml)并搅拌30分钟,混合物变得均匀。将所得溶液真空浓缩,然后加入吡啶(10ml)并真空除去三次。将所得固体溶于10ml吡啶中。缓慢加入二苯基磷酰氯(956μl,1当量),然而在反应混合物中形成沉淀,因此将其用另外的吡啶(50ml)稀释。1小时后,加入单(三丁胺)磷酸酯(3.3ml,3当量)并将其搅拌过夜。
在真空下除去溶剂,并将所得固体溶于水中并透析。透析后,通过冷冻干燥除去水,得到粘性固体。pnmr分析表明,87.7%的膦酸酯与相邻膦酸酯形成酸酐,而12.3%为膦酰基-单磷酸酯。
实施例36聚甲基-乙烯基膦酸盐的后聚合改性
按照与实施例32类似的过程,向磁力搅拌的干燥圆底烧瓶装入聚甲基-乙烯基膦酸盐、dmf并用氮气吹扫。接下来,加入三丁胺(相对于p单体为2.0当量)并在室温下搅拌过夜,得到均匀溶液。将cdi(相对于p单体为1.25当量)和dmf预混合并加入到搅拌过夜的溶液中。将h3po4(3当量)、三丁胺(3.5当量)和dmf混合并超声处理,然后加入到含聚合物的溶液中。将所得物在室温下搅拌过夜。在真空(9托)下除去所得溶液中的溶剂直至约60℃的最终温度,得到含膦酰基-磷酸根的聚合物。
实施例37含无规膦酸盐的聚合物的后聚合改性
膦酸化聚乙烯按照anbar(m.anbar、g.a.st.john和a.cscott,jdentres,第53卷,第4期,第867-878页,1974年)或schroeder和sopchak(j,p.schroeder和w.p.sopchak,journalofpolymerscience,第47卷,第149期,第417页,1960年)的描述合成。简而言之,使10g聚乙烯与200gpcl3在干燥烧瓶中回流反应,直至聚合物溶解。接下来,使干燥的氧气流过溶解的溶液。蒸馏所得溶液以使总体积减半,并将其倒在冰片上以形成膦酸化聚乙烯。按照实施例36的过程,使此膦酸化聚乙烯反应。
实施例38含聚(乙烯基苄基膦酸)的聚合物的后聚合改性
聚(乙烯基苄基膦酸)通过使用如anbar等人所述的在甲醇中加热使(4-乙烯基苄基)膦酸聚合来合成,或者通过使用引发剂诸如过硫酸铵(相对于单体为5-10%负载)来合成。按照实施例36的过程,使所得的聚合物反应。
实施例39侧链上膦酰基磷酸盐单体和聚合物的合成
按照实施例12的过程,用具有伯羟基的乙二醇二聚体、三聚体、四聚体或聚合物诸如二乙基(2-羟基乙氧基)乙氧基乙基膦酸盐代替二甲基(2-羟乙基)膦酸盐。膦酸乙酯封端的乙二醇单元可按照brunet等人(ernestobrunet、*
实施例40通过后聚合改性合成侧链上的膦酰基磷酸盐聚合物
如实施例38中所述使乙氧基化聚乙烯醇反应,以形成膦酸酯封端的乙氧基化聚乙烯醇聚合物。乙氧基化聚乙烯醇通过在85-120℃的温度和20-200psig的压力下在密封反应器中使聚乙烯醇在碱催化剂(诸如甲醇盐或氢氧化钠)作用下与在数小时内缓慢添加的环氧乙烷反应而合成。如实施例36中所述使该膦酸酯封端的聚合物反应以形成含膦酰基-磷酸根的聚合物,其中膦酰基-磷酸盐通过后聚合改性连接至该聚合物的侧链。
实施例41二甲基乙烯基膦酸盐(dmvp)与svs的共聚
将svs(aldrich,25%水溶液,11.0mmol)装入圆底烧瓶中,并且用流动氮气吹扫烧瓶的顶部空间15分钟。将烧瓶密封并加热至60℃保持15分钟。将过硫酸铵(aps,225mg)加入1.0g水中。在总共6小时内,每30分钟向反应中加入0.1mlaps溶液和0.1mldmvp(aldrich,1.5g,1.3ml,11.0mmol)。将所得物在60℃下搅拌24小时。
将粗反应溶液用250ml水稀释,并用2k分子量截留的渗析膜对反渗透水透析4天。透析水的初始ph为5.8,但降至2.5。
通过冷冻干燥从产物中除去水,得到2.4g白色固体。
基于nmr分析,聚合物包含得自svs的56.9摩尔%重复单元、43.1摩尔%的dmvp。水含量计算为10.4重量%。
实施例42(乙基膦酰基-单磷酸)-乙烯基醚和(amps)按实施例23的共聚进行的共聚
将(乙基膦酰基-单磷酸)-乙烯基醚(实施例11,4.7mmol)、水5ml和amps(4g50%的溶液,8.7mmol)加入圆底烧瓶中,并且用流动的氮气将烧瓶顶部空间吹扫15分钟。将烧瓶密封并加热至60℃保持15分钟,得到均匀溶液。将过硫酸铵(aps,306mg)溶于1.1g水中。在总共4小时内,每30分钟向反应中加入0.1mlaps溶液。将反应物在60℃下搅拌4小时。
将粗反应溶液用750ml水稀释,并用2k分子量截留的渗析膜对反渗透水透析8天。
通过冷冻干燥从产物中除去水,得到2.12g棕褐色固体。
基于nmr分析,聚合物包含得自amps的90.8摩尔%重复单元、得自(乙基膦酰基-单磷酸)-乙烯基醚的9.2摩尔%重复单元。发现该固体包含77%重量的水。
实施例43vpasvs共聚物上的pspm。
根据pspm模型测试来自实施例17的聚合物以及购自polysciencesinc.的聚乙烯基磺酸盐和聚乙烯基膦酸盐的均聚物。结果与焦磷酸盐和多磷酸盐一起在图1和表3(下文)中示出。
表3
实施例44vpasvs共聚物上的psrm。
根据psrm模型测试来自实施例17的聚合物以及购自polysciencesinc.的聚乙烯基磺酸盐和聚乙烯基膦酸盐的均聚物。结果与焦磷酸盐、多磷酸盐和水处理物一起在图2和表4(下文)中示出。
表4
实施例45vppsvs共聚物上的pspm。
根据pspm模型测试来自实施例20的聚合物。结果与焦磷酸盐和多磷酸盐一起示于图3和表5(下文)中。
表5
实施例46vppsvs共聚物上的psrm。
根据psrm模型测试来自实施例20的聚合物。结果与焦磷酸盐、多磷酸盐和水处理物一起示于图4和表6(下文)中。
表6
实施例47混合共聚物上的psrm和pspm。
根据psrm和pspm模型测试来自如下所述的先前实施例的聚合物。δl的结果与焦磷酸盐、多磷酸盐和水处理物一起示于下表7中。
表7
实施例48至52的一般化学方案-通过除去水来合成乙烯基膦酰基-单磷酸盐(vpp)或[乙烯基膦酸磷酸酐]和其他扩展的乙烯基膦酰基-磷酸盐(evpp)
以下化学方案显示了在实施例48至52中用于形成主要期望产物vpp和vppp的一般反应方案,连同在以下实验中的一些但不是全部中观察到的一些其他产物。有关最终确定的产物分布,请参考各个实施例。
实施例48-通过使用具有3当量的pa的吹扫气体进行蒸发来合成vpp和evpp
向50ml三颈圆底烧瓶中加入1克乙烯基膦酸(vpa)和2.72g(3当量)99%的磷酸(pa),该三颈圆底烧瓶配备有磁力搅拌器和位于中心颈的短程蒸馏头。一个侧颈被塞住,将氮气吹扫通过另一个侧颈并通过蒸馏头排出。将烧瓶置于加热至105℃的油浴中并在该温度下搅拌27小时。在期望的时间点取出样品(约1滴),溶解于具有0.25ml三丁胺的1mld7-dmf中,并且通过p-nmr评估。发现最终产物含有vpa、乙烯基膦酰基-单磷酸盐(vpp)、乙烯基-膦酰基-焦磷酸盐(vppp)、乙烯基-膦酸酐(vppv)、磷酸(pa)、焦磷酸(pp)和三磷酸(ppp)。使用lcms确认物质鉴定。此外,对最终27小时的样品进行h-nmr,由此确定在反应期间未发生聚合。
发现在27小时的熔体中所有含乙烯基的物质的最终摩尔分布为43%vpa、38%vpp、9%vppp和10%vppv。
实施例49-通过使用真空和具有3当量的pa的吹扫气体进行蒸发来合成vpp和evpp
按照实施例48的过程,其中改变如下。将短程蒸馏头连接到buchi真空泵,而不是排放到大气中。在实验期间,用恒定的氮气流从一个侧颈将圆底烧瓶抽空至50-60托。在32小时和48小时取样时,在含乙烯基的分布为31-32%vpa、40-41%vpp、14%vppp和13-14%vppv的时间点之间几乎没有变化。在p-nmr中也观察到了与vpppv相对应的信号,但没有被定量为与其他峰重叠。
实施例50-通过使用真空和具有6当量的pa的吹扫气体进行蒸发来合成vpp和evpp
按照实施例49的过程,使用相对于vpa为6当量的pa。在72小时含乙烯基物质的分布为31%vpa、40%vpp、21%vppp和8%vppv。在p-nmr中也观察到了与vpppv相对应的信号,但没有被定量为与其他峰重叠。
实施例51-通过与亚磷酸酐(p2o5,五氧化二磷)反应来合成vpp和evpp
向磁力搅拌的20ml闪烁小瓶中添加2.24g85重量%磷酸水溶液、1.01g90%乙烯基膦酸和2.5g五氧化二磷(按该顺序)。乙烯基膦酸盐与总磷酸盐的摩尔比(计算为磷酸盐摩尔数加上p2o摩尔数的两倍之和)为6。将小瓶加热至175℃并使用实施例x的过程在1小时时取样用于pnmr。所鉴定的含乙烯基物质的摩尔组成为34%vpa、41%vpp、19%vppp和5%vppv。另外的乙烯基峰在pnmr中可见,这些峰可能对应于较大的物质,包括vpppp和vppppp。lcms证实了更高级膦酰基-磷酸盐的存在,在负离子模式下,vpp、vppp、vpppp、vppppp、vpppppp和vppppppp的峰均可见。
实施例52-实施例49的放大和纯化
按照实施例49的过程,总材料增加了5倍。在32小时的采样中显示出含乙烯基物质的分布为35%vpa、37%vpp、12%vppp、12%vppv和4%vpppv。
冷却后,将大部分粗制反应混合物溶于40ml无水dmf中。将溶解的溶液加入到28.1g三乙胺(基于总的起始酸为1.5当量)的100ml无水dmf溶液中,同时快速搅拌5分钟。对所得溶液进行p-nmr,结果与粗反应混合物的分布一致。
在70℃和25托下从所得溶液除去dmf,得到38.4g粘稠黄色油状物。将其溶于100mlh2o中,得到ph为2.5的溶液,用110g10%naoh将ph调节至11.0,得到澄清溶液。对所得溶液进行p-nmr,显示出与先前样品一致的产物分布,但vppv降低了约20%。在室温下静置1小时后,形成白色沉淀,其通过过滤收集,在环境空气中干燥过夜至4.65克。发现该沉淀为约90%的焦磷酸盐,还有4%的磷酸盐和分别小于3%的vpa、vpp和ppp。除去滤液中的溶剂,得到49.4g透明粘稠油状物。用石蕊检查所得油状物的ph,发现其约为7。用额外的水使其达到大约125g,得到7.5的ph,再用15.2g1nnaoh调节至11.0。在室温下,在30分钟内,在快速搅拌下向该ph11溶液中加入250mlmeoh。在一小时内形成白色沉淀。通过过滤收集该沉淀,用50ml2:3h2o:meoh冲洗一次,并在环境空气下过夜干燥至17.9g。发现该沉淀为大约43%焦磷酸盐、39%磷酸、10%ppp、3%vpp和4%vppp。将meoh水溶液在室温下在流动氮气下浓缩过夜,得到31.1g粘稠油状物。发现该油状物具有大约33%vpa、33%vpp、8%vppp、11%pa、10%vppv和3%vpppv的摩尔磷分布。还发现该油状物具有残余的水和dmf。
在室温下经1小时向该油状物中加入300mlmeoh,得到白色沉淀,通过过滤收集该白色沉淀,用1x50mlmeoh冲洗并在室温下真空干燥2小时,得到4.3g白色粉末。发现该粉末具有49%vpp、26%pa、6%pp、15%vppp和3%vpa的摩尔磷分布。将meoh溶液在室温下在流动氮气下浓缩得到7.0g白色糊状物。发现白色糊状物的组成大约为73%vpa、23%vpp和5%vpppv。
实施例53-聚合以形成含有vppp的聚合物,并使用pspm和psrm进行测试
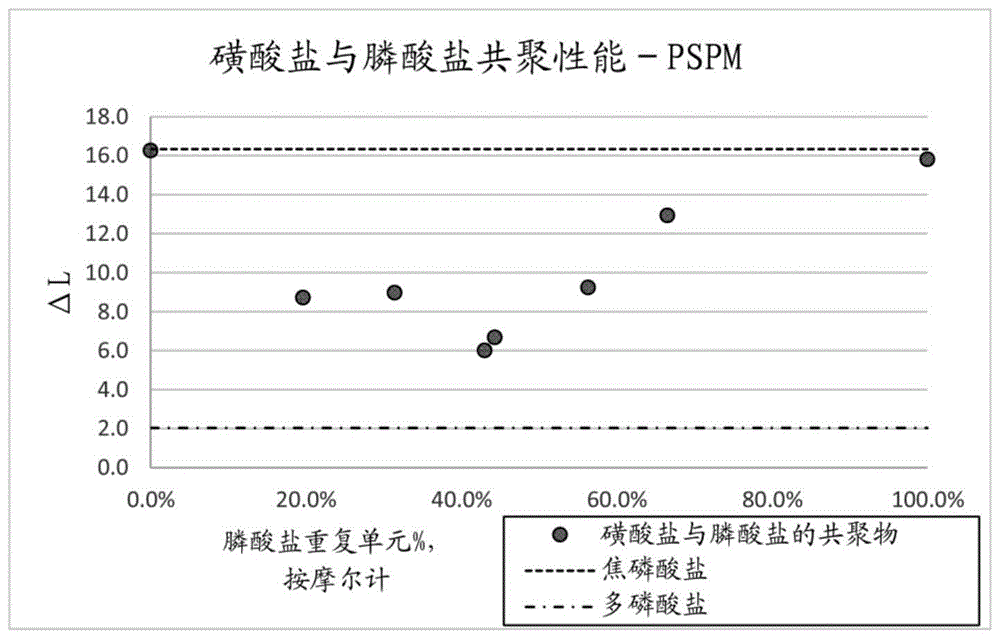
按照实施例19和20的过程,使用含有vppp的白色粉末(8.6mmol乙烯基)和svs(8.6mmol乙烯基)的50/50混合物(总摩尔乙烯基),将实施例52的含有49%vpp、26%pa、6%pp、15%vppp和3%vpa的白色粉末聚合。透析和冷冻干燥后,收集到3.6g聚合物,并且发现包含按svs计57%的单体,按膦酸盐计43%的单体。膦酸盐分布为3%来自vpa、78%来自vpp并且18%来自vppp。按重量计,聚合物为78%活性物质以及22%的杂质/水。在pspm和psrm模型中测试该聚合物,δl值分别为5.5和11.0。pspm的对照物为:水28.0、hap空白0.0、焦磷酸盐18.0、多磷酸盐4.0;以及psrm的对照物为:水24.2、hap空白0.0、焦磷酸盐12.4、多磷酸盐8.6。
实施例54至57-口腔护理制剂的放大和测试
下列实施例说明了将含有膦酰基磷酸盐的聚合物配制成洁齿剂,并随后在污渍模型中进行测试。
实施例54-实施例19和20的20g-30g放大比例
使用96.7mmol的vpp和96.7mmol的vsa以及同等增量的其他试剂和溶剂,放大实施例19和实施例20的过程。透析和冷冻干燥后,收集到27.1g聚合物,并且发现包含按svs计59%的单体、按vpp计40%的单体和按vpa计2%的单体。按重量计,聚合物为83%活性物质以及17%的杂质/水。
在pspm和psrm模型中测试该聚合物,δl值分别为6.8和13.0。pspm的对照物为:水28.0、hap空白0.0、焦磷酸盐14.3、多磷酸盐3.1;以及psrm的对照物为:水25.0、hap空白0.0、焦磷酸盐13.5、多磷酸盐10.7。
实施例55-实施例16和17的20g-30g放大比例
使用148mmol的vpp和122mmol的vsa以及同等增量的其他试剂和溶剂,放大实施例16和实施例17的过程。渗析和冷冻干燥后,收集到26.8g聚合物,并且发现包含按svs计54%的单体,按vpa计46%的单体。按重量计,聚合物为90%活性物质以及10%的杂质/水。在pspm和psrm模型中测试该聚合物,δl值分别为10.2和20.2。pspm的对照物为:水28.0、hap空白0.0、焦磷酸盐14.3、多磷酸盐3.1;以及psrm的对照物为:水25.0、hap空白0.0、焦磷酸盐13.5、多磷酸盐10.7。
实施例56-实施例19和20的100g放大比例
使用354.5mmol的vpp和433mmol的vsa以及同等增量的其他试剂和溶剂,放大实施例19和实施例20的过程。中和后,将本体溶液用水补充至9819g,并用1nnaoh将ph调节至10。使用tamiindustries1000mwco色谱柱(e190613n001),通过切向流过滤(tff)将所得溶液中的低分子量杂质降低。将溶液从贮存器中泵送通过色谱柱,然后再返回到贮存器中。将通过柱孔的流出物收集在天平上的烧瓶中。在第一次运行中,泵送溶液直至收集到3.5kg流出物。然后将贮存器中的剩余溶液补充回至约9kg。重复该过程,移除4.8kg并将贮存器补充至11kg。在最后一次运行中,移除6kg流出物。在最终tff后,使浓缩溶液通过0.22μm过滤器(stericup500ml过滤器单元,aldrich)过滤。
在室温下,在流动氮气下蒸发5天,过滤后,从最终的tff浓缩物中除去水,得到173克棕褐色糊状物。将其在>1托的真空下进一步干燥48小时,得到137.2g浅棕褐色固体。发现该固体含有基于svs的66%单体,基于vpp的34%单体。按重量计,聚合物为80%活性物质以及20%的杂质/水。在pspm和psrm模型中测试该聚合物,δl值分别为6.5和11.5。pspm的对照物为:水28.0、hap空白0.0、焦磷酸盐18.0、多磷酸盐4.0;以及psrm的对照物为:水24.2、hap空白0.0、焦磷酸盐12.4、多磷酸盐8.6。
实施例57-实施例54至56的制剂和测试
除非另外指明,否则本实施例中的所有百分比均按重量计。
如下制备组合物:
组合物#1是商购获得的crestcavityprotectionregularflavor。
组合物#2是商购获得的crestprohealthcleanmintsmoothformula。
组合物#3与加入了实施例54聚合物的组合物#2相同。
将组合物#2称重到speedmix广口瓶中。然后将实施例54聚合物加入到speedmix广口瓶中,并且在speedmixer中混合直至均匀。然后用ph电极测定ph,并且在speedmixer中加入2nhcl并混合,以将ph调节至目标值为约6。
组合物#4与加入了实施例55聚合物的组合物#2相同。将组合物#2称重到speedmix广口瓶中。然后将实施例55聚合物加入到speedmix广口瓶中,并且在speedmixer中混合直至均匀。然后用ph电极测定ph,并且在speedmixer中加入50%naoh溶液并混合,以将ph调节至目标值为约6。
在中试规模的混合器中,通过向混合器中加入约一半山梨醇,用槽罐上的加热/冷却夹套加热至65℃,并且抽真空,制备组合物#5。在单独的容器中,将1重量%的二氧化硅和所有羟乙基纤维素干混直至均匀,然后通过真空吸入混合容器中。锚式搅拌器和高剪切转子/定子设备均用于混合和均化混合物,以确保羟乙基纤维素的均质性和水化。一旦均匀,就关闭转子/定子设备。加入剩余的山梨醇、约25%的水和所有的蓝色染料,并使用锚式搅拌器混合直至均匀。在单独的容器中,将1重量%的二氧化硅、所有糖精和所有角叉菜胶干混,并且在高剪切转子/定子设备和锚式搅拌器运转的情况下真空吸入到主混合容器中。一旦均匀,就关闭转子/定子。接下来,将剩余的二氧化硅真空吸入主混合容器中,并使用锚式搅拌器在不小于26英寸汞柱的真空下混合。然后通过加热/冷却夹套将批料冷却至大约49℃,同时继续用锚式搅拌器混合。一旦批料达到49℃,停止锚式搅拌器,打开混合器并将风味剂和月桂基硫酸钠溶液添加到批料的顶部。然后抽真空至24英寸汞柱,并且开启锚式搅拌器和转子/定子,直至批料均匀混合。混合后,关闭转子/定子,并且抽真空至27英寸汞柱以除去空气。在单独的容器中,将剩余的75%的水加热至65℃。将葡萄糖酸钠加入到水中并混合直至溶解。然后将氟化亚锡加入到葡萄糖酸盐溶液中并混合直至溶解。然后将氯化亚锡加入到葡萄糖酸盐溶液中并混合直至溶解。一旦制备好该溶液,就将其在真空下加入到主混合容器中,并且使用锚式搅拌器混合直至均匀。混合后,在真空下将氢氧化钠加入到主混合容器中,并且使用锚式搅拌器和转子/定子均匀混合。一旦均匀,就关闭转子/定子,并且将加热/冷却夹套降低至30℃,并且抽真空至26英寸汞柱。将批料在真空下混合直至温度达到35℃,将其泵出主混合容器。
组合物#6与加入了实施例56聚合物的组合物#5相同。将组合物#5称重到speedmix广口瓶中。然后将实施例56聚合物加入到speedmix广口瓶中,并且在speedmixer中混合直至均匀。然后用ph电极测定ph,并且不需要进一步调节以实现约6的ph。
组合物#7与加入了实施例56聚合物的组合物#2相同。将组合物#2称重到speedmix广口瓶中。然后将实施例56聚合物加入到speedmix广口瓶中,并且在speedmixer中混合直至均匀。然后用ph电极测定ph,并且在speedmixer中加入50%naoh溶液并混合,以将ph调节至目标值为约6。
实施例58-通过与磷酸和脲反应合成vpp和evpp
对于实施例中的所有样品,遵循以下一般过程:
向闪烁小瓶中装入vpa、85%或99%h3po4、脲和水,如下表1所示。将所得物在60℃下搅拌大约15分钟,直到获得均匀的溶液。将所得溶液热转移到800ml烧杯中。将烧杯置于具有循环气流和外部通风的可编程实验室烘箱中。如下加热所有样品:
1)在15分钟内从室温升至110℃。
2)在110℃下保持3小时。
3)在15分钟内从110℃升至150℃。
4)如下表所示,在150℃下保持15或60分钟。
5)冷却至室温并使其静置过夜。
对粗反应产物(约50mg反应产物的1mld2o溶液,加5滴30%naod)进行p-nmr。发现这些产物含有vpa、乙烯基膦酰基-磷酸盐(vppa)、乙烯基膦酰基-焦磷酸盐(vpppa)、乙烯基膦酸酐(sm-an)、磷酸(pa)、焦磷酸和三磷酸(ppp)。来自p-nmr的面积示于下表3中。还对反应产物进行h-nmr,以检查加热期间vpa的聚合。未观察到聚合物。
实施例59-通过使聚合物与磷酸和脲的反应合成含有vpp和evpp的聚合物
将乙烯基膦酸二甲酯(dmvp,10.6g,77.9mmol)和乙烯基磺酸钠溶液(svs,25%水溶液,40.5g,77.9mmol)加入到100ml圆底烧瓶中。用氮气将烧瓶吹扫15分钟,并且加热至60℃。将888mg过硫酸铵(aps,占总单体的2.55%)加入4g水中,并用氮气脱气5分钟。将aps溶液加入到包含dmvp和svs的溶液中,并且使所得溶液在60℃氮气下搅拌24小时。
对粗反应溶液运行1h-nmr和31p-nmr,并且观察到单体转化率为约99%,在膦酸根基团的约37ppm处有宽p聚合物峰。
用207g水将粗反应溶液在水中稀释至10重量%的聚合物。向其中加入300ml丙酮,在室温下连续搅拌超过30分钟,得到浑浊溶液。在分液漏斗中静置30分钟后,形成下层粘稠富聚合物浆料和上层流体有机层。收集下层,溶剂在氮气下蒸发过夜,然后在1托下真空蒸发2小时,得到15.3g粘性棕褐色固体。用内标磷酸三甲酯对该固体运行1h-nmr和31p-nmr,显示dmvp:svs衍生基团的比率为50:50。
将粘性棕褐色固体与30g水和45g浓hcl(约等于37%)混合,得到乳白色溶液。将该混合物回流48小时,得到淡棕色的透明溶液。在60℃和20托下操作的旋转蒸发仪上将水和hcl从溶液中汽提至约等于20ml的总体积。再向该剩余馏分中加入100ml水并重复汽提,然后加入200ml水,将样品冷冻并冻干,得到11.8g棕褐色固体。31p-nmr显示聚合物峰从约等于37ppm偏移至约等于32ppm,而1h-nmr显示聚合物峰在约等于3.8ppm处消失,该处对应于甲酯峰。用内标分析表明,含p基团与含硫基团的比率为约47至53,并且重量活性为82.4%。
向100ml烧杯中加入4.85克85%的磷酸和2.77克脲,并且在60℃下加热15分钟,然后冷却至室温,得到澄清溶液。将5克82.4%的活性聚合物(p/s的计算比为47:53)溶于15ml水中,并将其添加到100ml烧杯中的磷酸/脲混合物中。将烧杯置于具有循环气流和外部通风的可编程实验室烘箱中并如下加热:
1)在15分钟内从室温升至110℃。
2)在110℃下保持3小时。
3)在15分钟内从110℃升至150℃。
4)在150℃下保持15分钟。
5)冷却至室温并使其静置过夜。
收集到11.4克海绵状白色产物。对粗反应产物(约150mg反应产物的1mld2o溶液,加2滴30%naod)进行p-nmr。p-nmr显示出约-5ppm处的宽峰,对应于聚合物链上的膦酰基-磷酸根基团。该峰的一部分与焦磷酸盐重叠,使得定量困难。
将大部分粗产物即11.4g溶于50ml水中,在搅拌下加入圆底烧瓶中,并在30分钟内加入50ml甲醇,得到浑浊溶液。在分液漏斗中静置30分钟后,得到较低粘度的富聚合物的浆料层,将其分离(9.5g)。通过p-nmr评估聚合物比磷酸盐比焦磷酸盐的比率,发现其为161比43比113。
使用50ml水和50ml甲醇对上述9.5g浆料重复沉淀。得到2.13g浆料。p-nmr显示聚合物比磷酸盐比焦磷酸盐的比率为158比3比18。
将所得浆料加至250ml反渗透(ro)水中,通过在thermoscientificslide-a-lyzer透析烧瓶(2kmwco)中用ro水(用碳酸氢钠饱和溶液将ph调节至8.5)透析6天来进一步纯化。通过冷冻和冻干除去水,得到1.59克白色固体。1h-nmr和31p-nmr显示所收集的聚合物约等于41%p单体和59%s单体。含p基团的分析显示约等于22%的膦酰基-磷酸根基团以及少量的膦酰基-焦磷酸根基团。剩余的含p基团似乎是膦酸盐和膦酸酯酸酐结构的混合物。聚合物计算为87.4重量%的活性物质。
实施例60乙烯基膦酰基-单磷酸盐(vpp)和乙烯基磺酸钠(svs)的共聚以及纯化
将vpp(如实施例1中所述以较大规模制备,64.6g活性物质,254mmol)和svs(25%水溶液,161.6g,310mmol)装入500ml圆底烧瓶中,其中svs与vpp的初始摩尔比为55至45,然后搅拌并用流动氮气吹扫烧瓶的顶部空间60分钟。通过加入9.5ml的1mnaoh,将溶液的ph从8.5升至10.5。将烧瓶用流动氮气吹扫并加热至60℃,此时加入过硫酸铵(aps,aldrich,7.73ml的10%水溶液,mg,相对于总单体为0.6%)。将所得物在60℃下搅拌24小时。
对粗反应溶液运行1h-nmr和31p-nmr。观察到总单体转化率为78%,膦酸根基团在约18ppm至23ppm处具有宽p聚合物峰,并且与膦酸根基团键合的磷酸根在-6ppm至-10ppm处具有宽p聚合物峰。
通过在15分钟内将甲醇等分试样加入到含有10%活性聚合物的搅拌溶液中来纯化该聚合物。得到浑浊溶液并将其转移到分液漏斗中,让其再静置15分钟以完全分离成下层粘稠富聚合物浆料和上层流体层。收集下层聚合物层,并使用额外的甲醇等分试样,然后重复相同的过程,使上层再沉淀。然后将所有样品在真空下干燥两天,最终质量记录在下表4中。
表4
此外,将约50ml剩余的上层h2o/meoh层在室温下在n2流下浓缩过夜,然后在室温下真空干燥24小时,得到2.5g白色固体。尺寸排阻色谱法/凝胶渗透色谱法(sec或gpc,3柱串联,polymerstandardsservicemcx1000a、mcx500a和mcx100a均为5μm,带有保护柱,0.2mnano3流动相1ml/min)显示分子量从分子量最高的馏分1到分子量最低的馏分6依次降低。由聚合物分析得到的gpc迹线图如图5所示。较高的分子量由较短的保留时间表示,而较低的分子量则具有较长的保留时间。22.5分钟后的大峰表示非聚合物物质,诸如残余单体和乙烯基硫酸钠溶液中的盐杂质。
实施例61得自实施例60的乙烯基膦酰基-单磷酸盐(vpp)和乙烯基磺酸钠(svs)样品的额外纯化
对馏分1-3和馏分4-6进行额外的纯化。由合并的馏分1-3形成15重量%的聚合物水溶液。在搅拌下向其中加入等于60分钟的全部水馏分质量的20%的甲醇质量。停止搅拌,并分离溶液相,得到富含粘性聚合物的下层。收集并干燥该馏分。将该过程再重复三次,每次加入相对于溶液起始质量额外的10%甲醇。所有样品均进行烘箱干燥,原始质量百分比记录在下表中。馏分1-4具有77-81%活性物质,还具有小于0.5%的磷酸盐、小于0.2%的乙烯基磷酸盐或乙烯基膦酰基磷酸盐,并且未检测到乙烯基磺酸盐。
表5
由合并的馏分4-6形成20重量%的聚合物溶液。向该溶液中加入等于溶液总质量的60%的甲醇质量。将所得的沉淀真空干燥两天。回收的聚合物质量为初始质量的93%,活性物质为83%,磷酸盐小于0.5%,乙烯基磺酸盐小于0.1%,并且乙烯基膦酸盐或乙烯基膦酸盐小于0.1%。
所得再分馏材料的gpc迹线图示于图6中。
除了ri检测,还进行了光散射。低保留时间的样品提供良好的光散射,而较高保留时间的样品则不能。此现象已通过未连接到gpc的独立光散射仪独立确认。低分子量馏分看起来聚集,这表现为在将光散射信号处理成分子量后的高分子量和高误差。为此,仅给出了保留较少的材料的分子量。对于保留较多的馏分,计算的mn和mw的增加趋势仍在继续,不确定度接近50%。检测再分馏馏分1-3-1中mw为60,000道尔顿的聚合物。这对应于250至450个重复单元的聚合物,具体取决于乙烯基磺酸盐和乙烯基膦酰基-磷酸盐衍生单元的组成。
表6
实施例62通过不同的分析技术鉴定端基
来自实施例61的再分馏样品的hnmr在6.5-5ppm的烯烃区域中显示宽聚合物峰。这些峰相对于4.0-1.0ppm的非烯烃峰的积分可用于估计存在多少烯烃。假设为乙烯类似基团,则将烯烃面积除以2,而假设为ch2-chx,则将非烯烃面积除以3,其中x为p或s。根据各馏分的组成,使用内标结合hnmr和pnmr计算,mn可在每种烯烃都对应于端基的假设下近似。然后,该计算与mn的光散射(ls)结果的接近度可用于衡量每种聚合物在末端位置是否具有烯烃。比较结果在表7中给出。
表7
对于保留较少且可能分子量较高的物质,匹配非常接近,再分馏馏分1-3-1和再分馏馏分1-3-2的值分别为5.6对6.8和3.5对2.7。与光散射不同,基于烯烃的分析还表明mn确实随着在柱上保留较多的馏分增加而减小。为了确认烯烃的ch2性质,对样品再分馏馏分4-6-1运行编辑的异核单量子相干(编辑的hsqc)nmr。证实烯烃峰具有ch2特征。
样品再分馏馏分1-3-4也通过离子色谱法(dionexionpacas16-4μm)然后通过高分辨率质谱法进行分析。除了具有许多信号的大的宽聚合物峰之外,还观察到尖锐的早期洗脱(较少的总电荷)峰。发现该早期洗脱峰含有膦酰基-磷酸根“三聚体”的多个质量并匹配该多个质量,包括质子形式、钠形式、质子和钠的混合物以及对应于水的损失或增加和磷酸根基团的损失的质量。发现该物质含有烯属或环状的不饱和基团。给定编辑的hsqc结果,示出了完全质子形式的结构。
假设也存在除烯烃之外的其他端基。在合成再分馏样品时,使用相对于可聚合单体的总摩尔数为0.6%的引发剂。典型的引发剂效率低于100%,但是为了计算起见,将使用该值。每种过硫酸盐均可分裂形成2个自由基。如果每个自由基开始一条聚合物链,并且反应进行至完成,则预计每条链具有83个重复单元。根据hnmr数据,初始的合并馏分1-3具有平均19个重复单元,而合并馏分4-6具有6个重复单元。因此,假设每个引发剂自由基完全引发聚合物,则在该链上至少发生4次链转移或回咬和β断裂组合,每次转移都可能产生烯烃。假设只有40%的引发效率,则预计将有208个重复单元,这意味着从每个引发剂自由基发生11次链转移或回咬和β断裂组合。
应当理解,本文所述的实施例和实施方案仅用于示例性目的,并且在不背离本申请的实质和范围的情况下,本领域专业人员可以对其做出各种变型或改变。
- 还没有人留言评论。精彩留言会获得点赞!