一种c-Myc基因表达抑制剂及其应用的制作方法
本发明涉及一种c-myc基因表达抑制剂及其应用。
背景技术:
聚酰胺一般是由2-3个不同的五元芳香杂环:n-甲基吡咯(py,芳香残基)、n-甲基咪唑(im,芳香残基)及3-羟基-n-甲基吡咯(hp,芳香残基)组成;在dna小沟中,二个聚酰胺或发卡聚酰胺反平行,边靠边与预先确定的dna碱基序列以1:1比例紧密、特异性识别碱基的合成小分子化合物,即n-甲基吡咯(py)倾向于识别t、a和c碱基;n-甲基咪唑(im)是g碱基的读取元素;3-羟基-n-甲基吡咯(hp)能特异性识别t碱基。到目前为止其中im/py和py/im配对分别识别g·c和c·g,而hp/py配对能把碱基对t·a、a·t区分开。而且聚酰胺具有良好的细胞渗透性,能够进入细胞核中,并且不易被核酸酶水解,相对转录因子能较强的键合特定靶基因。研究证明:它能通过在启动子区和增强子上干涉转录系统,调控与疾病相关突变基因的表达。
尽管人们利用聚酰胺中不同芳香残基的配对能将dna中的四个watson-crick碱基对特异性的区分开;但是,由五元芳香环组成的长链聚酰胺存在刚性过强,曲率过大,不能很好的与dna小沟曲率相匹配的不足;尤其是用hp作为t碱基的识别元素尚存在一些缺点。之前,本实验室成功研发利用(s)-β-羟基-γ-氨基酸残基(sγβ-oh)与β-丙氨酸残基(β)配对sγβ-oh/β能很好地将t·a、a·t、c·g、g·c区分开;而且,这样的氨基酸残基(sγβ-oh)具有柔韧性,起到‘弹簧’一样的作用,能够用于设计较长聚酰胺,使其有效识别人类基因中重复率较低的较长的dna序列,即解决使用较长聚酰胺识别较长dna序列的专一性问题。
因此,本实验室将在之前的基础上发明一种聚酰胺作为c-myc基因表达抑制剂,而且这种聚酰胺通过采用(s)-β-羟基-γ-氨基酸残基(sγβ-oh)与β-丙氨酸残基(β)配对(sγβ-oh/β)特异性识别碱基对t·a。
另一方面,20年之前,c-myc基因在人类伯基特淋巴瘤中发现,是禽类逆转录病毒的v-myc基因的细胞同源序列。在v-myc基因的研究基础上发现,c-myc基因是一种原癌基因,在许多癌症中表达出现异常。目前认为乳腺癌、结肠癌、宫颈癌、髓系白血病、黑色素瘤、骨肉瘤、胶质母细胞瘤及小细胞肺癌等恶性肿瘤都存在c-myc基因的失调、高表达。人c-myc基因位于8号染色体q24.1,包括两个内含子和三个外显子。
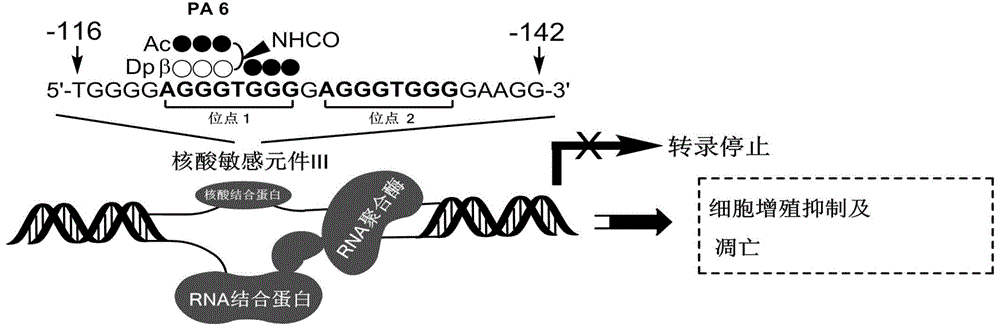
c-myc基因的转录由多个启动子调控。核酸超敏感元件iii1(nheiii1)也叫作pu27(27bp)控制c-myc基因80%~90%的转录活性。nheiii1富含鸟嘌呤(g),位于p1启动子上游-142bp~-115bp,能够形成具有转录活性的双螺旋结构。同时nheiii1在一定条件下可以形成分子内g-四链体结构,以无活性的沉默子结构形式抑制c-myc基因的转录。因此,nheiii1被认为通过下调c-myc基因的表达水平治疗肿瘤的潜在靶标。
c-myc原癌基因不仅调控15%的其他人类基因的表达,还参与调控细胞周期、新陈代谢、蛋白质生物合成和microrna调控等多种生理过程。除此之外,c-myc基因还参与调控细胞凋亡,细胞衰亡和dna损伤反应。通过活性氧和异常的dna复制中间体的生成,c-myc基因的过度表达会诱导产生dna损伤反应。dna损伤反应在肿瘤进展中具有双重作用,一方面能够抑制肿瘤的生长,而另一方面也能维护肿瘤的生长。因此,内源性c-myc基因具有内在矛盾性的特征。
c-myc在大多数人类干细胞中表达,并在正常和肿瘤细胞中都密切参与细胞周期、细胞分化、蛋白质合成和细胞凋亡的调控。因此,c-myc基因是癌症治疗发展中重要的基因靶标。目前以c-myc为靶标治疗肿瘤的小分子主要分为两类,一类是通过诱导并稳定c-myc基因启动子区域形成g-四链体来抑制肿瘤细胞的增殖;另一类是通过抑制c-myc/max二聚体的形成达到抑制肿瘤的作用。但是,到目前为止,人类还没有成功开发出以c-myc基因为靶标的临床抗肿瘤药物。
有关使用聚酰胺调控与疾病相关基因的报道:
2000年,chiangetal体外实验研究表明:聚酰胺有效抑制her-2/neu启动子区转录因子与dna的作用,从而抑制her-2/neu基因转录。2006年,matsudaetal首次应用小鼠动物模型研究表明:聚酰胺下调tgf-betal基因mrna及蛋白的表达,预示着聚酰胺有治疗tgf-betal基因高度表达的相关疾病的潜力,包括进展性肾炎、心瓣手术再狭窄。2010年,wang等人发现以鼠为动物模型研究聚酰胺下调mmp-9基因,能够抑制肿瘤的侵入和转移;表明mmp-9基因是聚酰胺抗肿瘤细胞转移的作用靶点。2007年,uenot等人报道利用聚酰胺调控结合组织生长因子ctgf基因。
通过以上对本发明相关的背景资料的检索分析发现:利用聚酰胺作为一种c-myc基因抑制剂;使用含有(s)-β-羟基-γ-氨基酸残基(sγβ-oh)的聚酰胺对c-myc启动子区序列特异性识别作用,从而抑制肿瘤细胞增殖,促进其凋亡,是有一定依据的。
技术实现要素:
针对现有技术存在的上述技术问题,本发明的目的在于提供一种c-myc基因表达抑制剂及其应用。
一种c-myc基因表达抑制剂,其特征在于包含吡咯-咪唑聚酰胺类化合物,所述吡咯-咪唑聚酰胺类化合物的发夹结构如pa1~pa6中的任意一种所示;
其中,●:n-甲基咪唑残基;○:n-甲基吡咯残基;
β表示β-丙氨酸残基;γ表示γ-氨基酸残基;
sγβ-oh表示(s)-β-羟基-γ-氨基酸残基;
ac表示乙酰基;dp表示n,n-二甲氨基丙二胺残基。
所述的一种c-myc基因表达抑制剂在制备抗肿瘤药物中的应用。
相对于现有技术,本发明取得的有益效果是:
自主设计合成了吡咯咪唑聚酰胺小分子,首次通过识别配对的方式靶向c-myc基因启动子序列,并且沉默c-myc基因的表达,其中发夹结构如pa6的吡咯-咪唑聚酰胺类化合物体现出较好的细胞毒性,并且能够抑制肿瘤细胞的生长,诱导肿瘤细胞凋亡。发夹结构如pa6的吡咯-咪唑聚酰胺类化合物抑制肿瘤细胞生长的具体过程如图1所示。
本发明的发夹结构如pa1~pa6的吡咯-咪唑聚酰胺类化合物与靶c-myc基因启动子区dna序列作用模式如图2所示。
附图说明
图1为本发明的聚酰胺抑制肿瘤细胞生长的过程示意图;
图2为本发明的聚酰胺与靶c-myc基因启动子区dna序列作用的模式图;
图3a为化合物pa1的制备路线图;
图3b为制备化合物pa1~pa6所使用的单体或二聚体的结构示意图;
图4为本发明的聚酰胺对a549细胞的生长抑制曲线图;
图5为本发明的聚酰胺对a549细胞的杀伤作用图;
图6为本发明的聚酰胺对sgc7901细胞的生长抑制曲线图;
图7为本发明的聚酰胺对sgc7901细胞的杀伤作用图;
图8a为本发明的聚酰胺对a549细胞的c-myc基因rt-pcr扩增产物电泳图;
图8b为本发明的聚酰胺对a549细胞内c-mycmrna水平的抑制图;
图8c为本发明的聚酰胺对a549细胞内c-myc基因蛋白免疫印迹试验产物电泳图;
图8d为本发明的聚酰胺对a549细胞内c-myc蛋白质水平的抑制;
图9a为本发明的聚酰胺对sgc7901细胞的c-myc基因rt-pcr扩增产物电泳图;
图9b为本发明的聚酰胺对sgc7901细胞内c-mycmrna水平的抑制图;
图9c为本发明的聚酰胺对sgc7901细胞内c-myc基因蛋白免疫印迹试验产物电泳图;
图9d为本发明的聚酰胺对sgc7901细胞内c-myc蛋白质水平的抑制;
图10为本发明的聚酰胺对a549细胞迁移的抑制作用图;
图11为本发明的聚酰胺对a549细胞迁移的抑制率图;
图12为本发明的聚酰胺对sgc7901细胞迁移的抑制作用图;
图13为本发明的聚酰胺对sgc7901细胞迁移的抑制率图;
图14a为本发明的聚酰胺诱导肿瘤细胞a549凋亡图之一;
图14b为本发明的聚酰胺诱导肿瘤细胞a549凋亡图之二;
图15a为本发明的聚酰胺诱导肿瘤细胞sgc7901凋亡图之一;
图15b为本发明的聚酰胺诱导肿瘤细胞sgc7901凋亡图之二;
图16a为本发明的聚酰胺阻滞a549细胞周期图之一;
图16b为本发明的聚酰胺阻滞a549细胞周期图之二;
图17a为本发明的聚酰胺阻滞sgc7901细胞周期图之一;
图17b为本发明的聚酰胺阻滞sgc7901细胞周期图之二。
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明的保护范围并不限于此。
实施例1:化合物pa1的制备
主要合成步骤:活化树脂、脱去树脂上保护基团、活化羧基基团、耦合、盖帽、切割树脂等部分。
活化树脂:将树脂用dmf进行充分溶胀。
脱保护:将fmoc保护基团从fmoc保护的树脂上通过哌啶/dmf溶液去保护,释放出游离氨基。
活化羧基:将参与下一个耦合反应的氨基酸羧基部分通过活化试剂hbtu进行活化,形成活化酯,从而高效的与氨基耦合。
耦合:活化好氨基酸与树脂上游离氨基反应,形成肽键。
盖帽:在反应中存在部分未完全反应的氨基结构,必须进行封盖,避免在下次耦合中引起副反应,从而保证合成目标聚酰胺的纯度。
切割:当目标聚酰胺合成结束,需要用dp作为切割试剂将其从树脂上切割下来。
所使用的初始原料和试剂试剂的缩写如表1。
表1:固相合成反应中所用试剂及其缩写
步骤如下:合成步骤如图3a。
1)活化:称取图3b如式(12)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3b中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3b中如式(4)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3b中如式(5)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)的操作,对步骤12)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
14)偶合:称取图3b中如式(7)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
15)重复步骤4)、步骤2)的操作,对步骤14)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
16)偶合:称取图3b中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
17)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤16)的反应液进行同样的盖帽、脱保护、盖帽反应;获得中间体(pa1-a);
为了验证合成产物,进行与步骤4)相应过程相同。
18)将步骤17)的中间体,连有目标聚酰胺(pa1)的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,得到手性羟基保护的发夹聚酰胺(pa1)的中间体(pa1-b);
19)在氮气保护下,向步骤18)的中间体加入3ml脱保护试剂重量配比为91:3:3:3的tfa/tms/tis/h2o混合液,室温反应1h脱去羟基上的叔丁基保护,真空下移除溶剂,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤得到白色的目标产物,含手性(s)-β-羟基-γ-氨基酸修饰的发夹聚酰胺(pa1)。收率15.6%,hplc97.6%,1h-nmr(400mhz,dmso-d6):10.42(s,1h,芳香nh),10.38(s,1h,芳香nh),10.14(s,2h,芳香nh),9.81(s,1h,芳香nh),9.67(br,2h,芳香nh),8.04-7.47(m,3h,脂肪nh),7.83-7.71(m,2h,脂肪nh),7.64(s,1h,芳香ch),7.50(s,1h,芳香ch),7.58(s,1h,芳香ch),7.48(s,1h,芳香ch),7.44(m,2h,芳香ch),7.39(s,1h,芳香ch),7.27(m,2h,芳香ch),5.53(br,1h,oh),3.98(m,1h,hoch),3.94(s,6h,2×nch3),3.90(s,3h,nch3),3.86(s,6h,2×nch3),3.80(s,3h,nch3),3.64(m,2h,nhch2),3.51(m,2h,nhch2),3.50-3.41(m,2h,nhch2),3.32-3.25(m,4h,2×nhch2),2.68(m,2h,nme2ch2),2.46(d,3jh,h=7.2hz,2h,coch2),2.33(t,3jh,h=6.8hz,2h,coch2),2.25-2.20(m,4h,2×coch2),2.02(s,6h,n(ch3)2),1.96(s,1h,coch3),1.76(m,2h,chch2ch2),1.51(m,2h,ch2ch2ch2)。esi-mass:m/zcalcdforc54h74n21o12[m+h]+:1208.58;found:1208.47[m+h]+。
实施例2:化合物pa2的制备
合成步骤类似合成pa1(图3a)。步骤如下:
1)活化:与制备pa1第一步1)活化操作相同。称取图3b如式(12)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3b中如式(8)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3b中如式(5)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3b中如式(7)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)的操作,对步骤12)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
14)偶合:称取图3b中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
15)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤14)的反应液进行同样的盖帽、脱保护、盖帽反应;获得中间体(pa2-a);
为了验证合成产物,进行与步骤4)相应过程相同。
16)将步骤15)的中间体,连有目标聚酰胺(pa2)的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,得到手性羟基保护的发夹聚酰胺(pa2)的中间体(pa2-b);
17)在氮气保护下,向步骤16)的中间体加入3ml脱保护试剂重量配比为91:3:3:3的tfa/tms/tis/h2o混合液,室温反应1h脱去羟基上的叔丁基保护,真空下移除溶剂,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤得到淡白色的目标产物,含手性(s)-β-羟基-γ-氨基酸修饰的发夹聚酰胺(pa2)。收率13.6%,hplc98.1%,1h-nmr(400mhz,dmso-d6):10.23(s,1h,芳香nh),10.01(s,1h,芳香nh),9.90-9.98(br,2h,芳香nh),9.75(s,1h,芳香nh),9.86(br,2h,芳香nh),8.15-7.89(m,3h,脂肪nh),7.63-7.41(m,2h,脂肪nh),7.39-7.35(br,2h,芳香ch),7.30(s,1h,芳香ch),7.27(s,1h,芳香ch),7.16(s,1h,芳香ch),7.13(m,2h,芳香ch),7.10(m,2h,芳香ch),5.52(br,1h,oh),3.99(m,1h,hoch),3.91(s,6h,2×nch3),3.87(s,3h,nch3),3.83(s,6h,2×nch3),3.80(s,3h,nch3),3.76(s,6h,2×nch3),3.72(s,3h,nch3),3.64(m,2h,nhch2),3.50(m,2h,nhch2),3.46-3.40(m,2h,nhch2),3.34-3.20(m,4h,2×nhch2),2.46(d,3jh,h=7.2hz,2h,coch2),2.36(t,3jh,h=6.8hz,2h,coch2),2.28-2.23(t,3jh,h=6.8hz,2h,coch2),2.01(s,6h,n(ch3)2),1.95(s,1h,coch3),1.79(m,2h,chch2ch2),1.53(m,2h,ch2ch2ch2)。esi-mass:m/zcalcdforc57h75n22o12[m+h]+:1259.59;found:1259.62[m+h]+。
实施例3:化合物pa3的制备
合成步骤类似合成pa1(图3a)。步骤如下:
1)活化:与制备pa1第一步1)活化操作相同。称取图3b如式(12)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3b中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3b中如式(7)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3b中如式(5)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)的操作,对步骤12)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
14)偶合:称取图3b中如式(4)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
15)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
16)偶合:称取图3b中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
17)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤16)的反应液进行同样的盖帽、脱保护、盖帽反应;获得中间体(pa3-a);
为了验证合成产物,进行与步骤4)相应过程相同。
18)将步骤17)的中间体,连有目标聚酰胺(pa3)的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,得到手性羟基保护的发夹聚酰胺(pa3)的中间体(pa3-b);
19)在氮气保护下,向步骤18)的中间体加入3ml脱保护试剂重量配比为91:3:3:3的tfa/tms/tis/h2o混合液,室温反应1h脱去羟基上的叔丁基保护,真空下移除溶剂,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤得到淡白色的目标产物,含手性(s)-β-羟基-γ-氨基酸修饰的发夹聚酰胺(pa3)。收率16.1%,hplc98.3%,1h-nmr(400mhz,dmso-d6):10.40(s,1h,芳香nh),10.37(s,1h,芳香nh),10.12(s,2h,芳香nh),9.80(s,1h,芳香nh),9.69(br,2h,芳香nh),8.08-7.45(m,3h,脂肪nh),7.81-7.70(m,2h,脂肪nh),7.66(s,1h,芳香ch),7.52(s,1h,芳香ch),7.57(s,1h,芳香ch),7.49(s,1h,芳香ch),7.41(m,2h,芳香ch),7.36(s,1h,芳香ch),7.26(m,2h,芳香ch),5.52(br,1h,oh),3.97(m,1h,hoch),3.93(s,6h,2×nch3),3.91(s,3h,nch3),3.87(s,6h,2×nch3),3.82(s,3h,nch3),3.63(m,2h,nhch2),3.50(m,2h,nhch2),3.48-3.40(m,2h,nhch2),3.31-3.25(m,4h,2×nhch2),2.69(m,2h,nme2ch2),2.45(d,3jh,h=7.2hz,2h,coch2),2.36(t,3jh,h=6.8hz,2h,coch2),2.26-2.20(m,4h,2×coch2),2.01(s,6h,n(ch3)2),1.94(s,1h,coch3),1.76(m,2h,chch2ch2),1.50(m,2h,ch2ch2ch2)。esi-mass:m/zcalcdforc54h74n21o12[m+h]+:1208.58;found:1208.53[m+h]+。
实施例4:化合物pa4的制备
合成步骤类似合成pa1(图3a)。步骤如下:
1)活化:与制备pa1第一步1)活化操作相同。称取图3b如式(12)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3b中如式(7)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3b中如式(5)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2),对步骤10)的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3b中如式(10)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)重复步骤4)、步骤2)和步骤4)的操作,分别对步骤14)的反应液进行同样的盖帽、脱保护、盖帽反应;获得中间体(pa4-a);
为了验证合成产物,进行与步骤4)相应过程相同。
14)将步骤13)的中间体,连有目标聚酰胺(pa4)的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,得到手性羟基保护的发夹聚酰胺(pa4)的中间体(pa4-b);
15)在氮气保护下,向步骤14)的中间体加入3ml脱保护试剂重量配比为91:3:3:3的tfa/tms/tis/h2o混合液,室温反应1h脱去羟基上的叔丁基保护,真空下移除溶剂,二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤得到淡白色的目标产物,含手性(s)-β-羟基-γ-氨基酸修饰的发夹聚酰胺(pa4)。收率11.4%,hplc98.3%,1h-nmr(400mhz,dmso-d6):10.20(s,1h,芳香nh),10.00(s,1h,芳香nh),9.92-9.99(br,2h,芳香nh),9.76(s,1h,芳香nh),9.87(br,2h,芳香nh),8.17-7.89(m,3h,脂肪nh),7.65-7.42(m,2h,脂肪nh),7.39-7.33(br,2h,芳香ch),7.35(s,1h,芳香ch),7.28(s,1h,芳香ch),7.16(s,1h,芳香ch),7.11(m,2h,芳香ch),7.06(m,2h,芳香ch),5.51(br,1h,oh),3.98(m,1h,hoch),3.90(s,6h,2×nch3),3.86(s,3h,nch3),3.82(s,6h,2×nch3),3.79(s,3h,nch3),3.76(s,6h,2×nch3),3.71(s,3h,nch3),3.66(m,2h,nhch2),3.51(m,2h,nhch2),3.44-3.38(m,2h,nhch2),3.32-3.22(m,4h,2×nhch2),2.44(d,3jh,h=7.2hz,2h,coch2),2.34(t,3jh,h=6.8hz,2h,coch2),2.27-2.21(t,3jh,h=6.8hz,2h,coch2),2.00(s,6h,n(ch3)2),1.96(s,1h,coch3),1.79(m,2h,chch2ch2),1.52(m,2h,ch2ch2ch2)。esi-mass:m/zcalcdforc57h75n22o12[m+h]+:1259.59;found:1259.55[m+h]+。
实施例5:化合物pa5的制备
合成步骤类似合成pa1(图3a)。步骤如下:
1)活化:与制备pa1第一步1)活化操作相同。称取图3b如式(12)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3b中如式(8)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3b中如式(8)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3b中如式(7)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应。
为了验证合成产物,进行与步骤4)相应过程相同。
10)重复步骤3)、步骤4)、步骤2),对固相反应管中的反应物进行偶合、盖帽和脱保护反应。
为了验证合成产物,进行与步骤4)相应过程相同。
11)偶合:称取图3b中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
12)重复步骤4)、步骤2)的操作,对步骤11)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
13)偶合:称取图3b中如式(6)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
14)重复步骤4)、步骤2)的操作,对步骤13)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
15)偶合:称取图3b中如式(11)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
16)重复步骤4)、步骤2)的操作,对步骤15)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
17)偶合:称取图3b中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
18)重复步骤4)、步骤2)的操作,对步骤17)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
19)偶合:称取图3b中如式(4)所示的单体(1.6mmol)(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
20)重复步骤4)、步骤2)的操作,对步骤19)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
21)重复步骤15)、步骤4)、步骤2)的操作,对步骤20)中的相反应管中的反应物进行偶合、盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
22)重复步骤15)、步骤4)、步骤2)和步骤4)的操作,分别对步骤21)的反应液进行同样的偶合、盖帽、脱保护、盖帽反应;获得中间体(pa5-a);
为了验证合成产物,进行与步骤4)相应过程相同。
23)将步骤22)的中间体(pa5-a),连有目标聚酰胺的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp,得到手性羟基和氨基保护的发夹聚酰胺(pa5)的中间体(pa5-b);
24)在氮气保护下,向步骤23)的中间体加入3ml脱保护试剂重量配比为91:3:3:3的tfa/tms/tis/h2o混合液,室温反应1h脱去羟基上的叔丁基和氨基上的叔丁氧羰基保护,真空下移除溶剂;二氯甲烷溶解产物,用5mol/l的盐酸调至酸性,有固体析出,过滤得到淡白色的目标产物,含手性(s)-β-羟基-γ-氨基酸修饰的发夹聚酰胺(pa5)。收率10.9%,hplc97.3%。1h-nmr(400mhz,dmso-d6):δ10.35(s,1h,芳香nh),10.31(s,1h,芳香nh),10.27(s,2h,芳香nh),10.22(s,1h,芳香nh),10.16(s,2h,芳香nh),9.98(s,1h,芳香nh),9.94(s,2h,芳香nh),9.88(s,1h,芳香nh),9.80(s,2h,芳香nh),9.86(s,1h,芳香nh),8.23-7.87(m,3h,脂肪nh),7.82-7.75(m,2h,脂肪nh),7.66(s,1h,芳香ch),7.59(s,1h,芳香ch),7.52(m,2h,芳香ch),7.47(s,1h,芳香ch),7.40-7.34(m,2h,芳香ch),7.30(s,2h,芳香ch),7.26-7.21(m,3h,芳香ch),7.17-7.00(m,4h,芳香ch),6.94-6.90(m,2h,芳香ch),6.86-6.80(m,4h,芳香ch),5.50(br,1h,oh),3.95(m,1h,hoch),3.91(s,3h,nch3),3.89(s,3h,nch3),3.87(s,6h,2×nch3),3.84(s,3h,nch3),3.79(s,6h,2×nch3),3.76(s,6h,2×nch3),3.72(s,3h,nch3),3.68(s,6h,2×nch3),3.64(s,6h,2×nch3),3.60(m,2h,nhch2),3.54(m,2h,nhch2),3.48-3.40(m,3h,nhch2+ch2ch2chnh3),3.34-3.20(m,4h,2×nhch2),2.63(m,2h,nme2ch2),2.45(d,3jh,h=7.4hz,2h,coch2),2.35(t,3jh,h=7.2hz,2h,coch2),2.29-2.23(m,2h,coch2),2.01(s,6h,n(ch3)2),1.96(s,1h,coch3),1.77(m,2h,chch2ch2),1.53(m,2h,ch2ch2ch2)。esi-mass:m/zcalcdforc98h120n42o20[m+2h]2+:2204.97;found:1102.47[m+2h]2+.
实施例6:化合物pa6的制备
合成步骤类似合成pa1(图3a)。步骤如下:
1)活化:与制备pa1第一步1)活化操作相同。称取图3b如式(12)所示的fmoc保护β-丙氨酸-clear树脂(sps-1,1.00g,0.4mmol,peptidesinternational)加入到固相反应装置的固相反应管中,通氮气保护,再加入5ml的dmf,使氮气充分鼓泡30min进行活化树脂;
2)脱保护:先配制3ml的20%(v/v)哌啶/dmf(哌啶、dmf均为处理过的无水溶剂),在氮气保护下加入到步骤1)的固相反应管,充分鼓泡反应15min,脱除β-丙氨酸上的氨基保护基团,抽去反应管中的溶剂,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
3)偶合:称取图3b中如式(8)所示的二聚体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入另一支无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
4)盖帽:配制5ml的5%乙酸酐+5%吡啶/dmf溶液,氮气保护下加入到固相反应管中进行盖帽15min,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂。
这一步为了验证合成产物,从固相反应管中取固体树脂(5mg)加入到5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液进行hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
5)重复步骤2),进行脱保护;
6)偶合:称取图3b中如式(2)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化。再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
7)重复步骤4)、步骤2),对步骤6)中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行完步骤4)后,从该固相反应管中分别取固体树脂(5mg)加入5ml的反应瓶中,加入500μl的dp,反应液于55℃反应过夜,切除树脂载体上的产物,用0.45μm的一次性过滤器过滤,取25μl的样品液金星hplc分析(254nm,0.1%tfa水溶液和乙腈为流动相,梯度洗脱,乙腈从0%到100%,流速1.0ml/min)。
8)偶合:称取图3b中如式(6)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,分别加入5ml无水dmf,振摇至化合物完全溶解,再分别加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
9)重复步骤4)、步骤2),对固相反应管中的反应物进行盖帽和脱保护反应。
为了验证合成产物,进行与步骤4)相应过程相同。
10)偶合:称取图3b中如式(11)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入无水反应瓶,加入5ml无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液分别加入固相反应管中,氮气鼓泡反应2.5h,抽干反应液,用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
11)重复步骤4)、步骤2)的操作,对步骤10)中的相反应管中的反应物进行盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
12)偶合:称取图3b中如式(3)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
13)依次重复步骤4)、步骤2)和步骤4)的操作,分别对步骤12)的反应液进行同样的盖帽、脱保护、盖帽反应;获得中间体(pa6-a)。
为了验证合成产物,进行与步骤4)相应过程相同。
14)向步骤13)所获得的中间体,连有目标聚酰胺的树脂中加入脱保护试剂重量配比为10:90的tfa/ch2cl2混合液后,室温氮气鼓泡反应0.5h,抽干反应液,分别无水二氯甲烷、无水dmf淋洗,每次淋洗后都抽干溶剂,获得脱去氨基上的叔丁氧羰基的发夹聚酰胺(pa6)的中间体(pa6-b);
15)重复步骤10)、步骤4)、步骤2)的操作,对步骤14)中的相反应管中的反应物进行偶合、盖帽和脱保护反应;
为了验证合成产物,进行与步骤4)相应过程相同。
16)偶合:称取图3b中如式(1)所示的单体(1.6mmol)和hbtu(576.5mg,1.52mmol)加入到无水反应瓶中,加入5ml等量无水dmf,振摇至化合物完全溶解,再加入1ml的diea,继续振摇反应20分钟至羧基完全活化,再将溶液加入到固相反应管中,氮气鼓泡反应2.5h,抽干反应液,分别用3ml无水二氯甲烷淋洗两次,2ml无水dmf淋洗一次,每次淋洗后都抽干溶剂;
17)重复步骤4),对步骤16)的反应液进行同样的盖帽反应;获得发夹聚酰胺(pa6)中间体的树脂(pa6-c)。
为了验证合成产物,进行与步骤4)相应过程相同。
18)将步骤17)的中间体,连有目标聚酰胺(pa6)的树脂从固相反应管中取出,放入反应瓶中,加入20ml的dp,油浴中55℃过夜反应,停止反应,过滤,收集滤液,真空下除去dp后,得到手性羟基保护的发夹聚酰胺(pa6)的中间体(pa6-d);
19)在氮气保护下,用二氯甲烷溶解步骤18)产物,用5mol/l的盐酸调至酸性,有固体析出,过滤得到白色的目标产物(pa6)。收率12.8%,hplc98.6%,1h-nmr(400mhz,dmso-d6):δ10.56(s,1h,芳香nh),10.47(s,1h,芳香nh),10.35(s,1h,芳香nh),10.23(s,2h,芳香nh),9.14(s,1h,芳香nh),9.90(s,1h,芳香nh),9.76(brs,1h,芳香nh),8.44-8.31(m,1h,脂肪nh),7.88-7.76(m,3h,脂肪nh),7.63(s,1h,芳香ch),7.57(s,1h,芳香ch),7.52(s,1h,芳香ch),7.46(m,2h,芳香ch),7.40(s,1h,芳香ch),7.34(m,1h,芳香ch),7.27(s,1h,芳香ch),7.14-7.10(m,3h,芳香ch),6.98-6.96(m,2h,芳香ch),3.92(s,6h,2×nch3),3.89(s,3h,nch3),3.86(s,6h,2×nch3),3.80(s,3h,nch3),3.75(s,3h,nch3),3.70(s,3h,2×nch3),3.61(m,2h,nhch2),3.56(m,2h,nhch2),3.38-3.26(m,3h,nhch2+ch2ch2chnh3),2.62(m,2h,nme2ch2),2.31(t,3jh,h=6.8hz,2h,coch2),2.01(s,6h,n(ch3)2),1.99(s,1h,coch3),1.79(m,2h,chch2ch2),1.47(m,2h,ch2ch2ch2)。esi-mass:m/zcalcdforc62h77n28o12[m+h]+:1405.63;found:1405.58[m+h]+.
实施例7:聚酰胺对c-myc基因高表达的肿瘤细胞的生长抑制
用rtca技术考查六种聚酰胺对c-myc基因高表达的a549和sgc7901两种肿瘤细胞的细胞毒性,以筛选出几种对肿瘤细胞毒性强的聚酰胺,获得较低ic50值(使细胞50%致死的药物浓度)的预选药物分子。
实验材料与方法(一)
材料:
a.化合物:发夹结构如pa1~pa6的吡咯-咪唑聚酰胺类化合物
b.细胞株:a549(人肺腺癌细胞),sgc-7901(胃低分化腺癌细胞)。
c.培养基组成:rpmi1640培养基(invitrogengibco,美国),小牛血清(invitrogengibco,美国),青霉素-链霉素双抗(invitrogengibco,美国),l-谷氨酰胺(invitrogengibco,美国),肝素(invitrogengibco,美国)。
肿瘤细胞的培养和收集:从液氮罐中取出肿瘤细胞,迅速置于水浴锅内,并不停摇动使其尽量融化。将解冻后的肿瘤细胞转移到离心管中,补充rpmi-1640全培养基至合适体积,混匀,低温低速离心除去上清液。重复使用全培养液洗涤一次,再用培养基悬浮细胞,按细胞密度接种至含全培养基的培养瓶中。至于合适的温度、co2、湿度的培养箱中培养。待细胞铺满培养瓶底,用胰酶消化为单个细胞,并转移到离心管中,补充全培养基,混匀,低温低速离心,除去上清液,重复洗涤一次,收集细胞沉淀。
实验方法:
rtca实验:e-plate实验所用的孔内加入全培养基,其余孔及所有孔间间隙中加入pbs溶液,室温孵育后,加入细胞稀释液,使每个孔的细胞数大约为5000-10000个,室温孵育后,取出e-plate,于实验孔内加入预先配置好的不同浓度的化合物,每个浓度设置一个重复孔。0.5%dmso作为对照;”0”只有培养液,为空白对照。
聚酰胺pa1~pa6对a549细胞的生长抑制曲线如图4所示。
对比6种聚酰胺药物对c-myc基因高表达的a549(肺腺癌)细胞的生长抑制作用,由图4中可以看出聚酰胺pa1、pa3、pa5和pa6对a549细胞的抑制作用都比较明显,尤其聚酰胺pa5和pa6有较好的浓度依赖性,其余2种聚酰胺药物只有当浓度达到一定值时,实验条件是16μm时,对靶细胞的抑制作用明显增强。
聚酰胺pa1~pa6对a549细胞的杀伤作用:通过数据分析加入聚酰胺pa1~pa6处理24小时后a549细胞的存活率如图5和表2所示。
图5显示了在加入聚酰胺pa1~pa6处理24小时后对a549的杀伤作用。对比这6种聚酰胺pa1~pa6在24小时内对a549杀伤作用,其中聚酰胺pa1~pa6的使用浓度为1μm,2μm,4μm,8μm,16μm。六种聚酰胺pa1~pa6在24小时内对a549细胞的杀伤作用基本上呈浓度依赖关系,其中聚酰胺pa5和pa6在16μm时的细胞的存活率仅分别为31.05%和7.14%,而聚酰胺pa2和pa4对细胞的杀伤作用比较弱,分别为51.55%和72.64%。综合图5的结果,聚酰胺pa5和pa6对a549的毒性作用较强,尤其是聚酰胺pa6;而聚酰胺pa4在实验浓度下对a549细胞没有明显的抑制作用。
六种聚酰胺pa1~pa6对c-myc基因高表达的sgc7901(肺腺癌)细胞的生长抑制作用,实验材料与方法同上述实验材料与方法,实验结果如图6所示。
从图6中可以看出聚酰胺pa1、聚酰胺pa3、聚酰胺pa5、聚酰胺pa6对sgc7901细胞呈现良好的细胞毒性,在浓度为16μm时,细胞指数曲线基本持平,说明sgc7901细胞的生长受到强烈的抑制。而聚酰胺pa2和聚酰胺pa4在8μm时,细胞生长并没有受到明显抑制。
聚酰胺对sgc7901细胞的杀伤作用:通过数据分析加入化合物pa1~pa6处理24小时后sgc7901细胞的存活率如图7和表2所示。
图7显示了在加入聚酰胺pa1~pa6处理24小时后sgc7901的存活率,在最大浓度16μm下,聚酰胺pa5和pa6对sgc7901细胞的存活率均在40%以下,而聚酰胺pa2和pa4存活率均在50%以上。经过对比得出聚酰胺pa1、pa3、pa5和pa6对sgc7901细胞的抑制作用都比较明显,而浓度依赖性较好的是聚酰胺pa5和pa6,其余4种聚酰胺都是当浓度增大到16μm时,对细胞的抑制作用明显增强。
rtca实验表明六种聚酰胺在不同程度上都促进a549细胞和sgc7901细胞的凋亡,其中聚酰胺pa5和pa6的作用效果比较好,聚酰胺pa6比pa5的作用强;对sgc7901细胞作用效果更好。这说明聚酰胺具有靶向基因、治疗多种与c-myc基因相关的肿瘤的潜力。
表2:聚酰胺1-6处理的a549和sgc7901细胞系的ic50值(μm)。
实施例8:聚酰胺对肿瘤细胞c-myc基因表达的影响
(1)通过rt-pcr技术,考查6种聚酰胺pa1~pa6对a549,sgc7901肿瘤细胞c-myc基因的mrna表达情况。
实验材料与方法(二)
材料:核酸提取试剂盒(rneasyminikit试剂盒,qiagen,德国),onesteprnapcrkit试剂盒(rr024b,takara,日本)试剂盒,胰酶(invitrogen,美国),bca蛋白浓度测定试剂盒(上海碧云天生物技术公司)。
培养基的组成:同实验材料与方法(一)
肿瘤细胞的培养与收集:同实验材料与方法(一)
rna的提取:rna提取完全参照rneasyminikit(qiagen说明书)。每种肿瘤细胞分别提取两次,提取完成后测定rna浓度并用depc处理过后调节合适浓度。
rt-pcr反应:rt-pcr反应采用onesteprnapcrkit(takara,rr024b)试剂盒。进行两次独立实验。
1.5%琼脂糖凝胶电泳,各取同样体积的rt-pcr反应产物,电泳30-60min,在紫外凝胶成像系统进行拍照,用图像分析软件进行数据分析。
(2)蛋白免疫印迹试验,考查6种聚酰胺pa1~pa6对a549,sgc7901肿瘤细胞c-myc基因的蛋白质表达情况。
表3:sds-聚丙烯酰胺凝胶配方
sds-聚丙烯酰胺凝胶凝电泳条件:sds-聚丙烯酰胺凝胶按表3制备;接通电源;在上样孔中加入maker和样品浓缩胶恒压80-100v,分离胶恒压100-140v,至溴酚蓝的条带刚跑出分离胶前沿后,停止电泳,然后,进行数据分析。
聚酰胺pa1~pa6分别加入a549,sgc7901细胞内,c-mycmrna水平的表达情况:在a549细胞内如图8a和图8b所示,在sgc7901细胞内如图9a和图9b所示,β-actin基因为对照基因。图8a和图9a为c-myc基因rt-pcr扩增产物电泳图。c-myc蛋白水平的表达情况:在a549细胞内如图8c和图8d所示,在sgc7901细胞内如图9c和图9d所示,β-actin基因为对照基因,图8c和图9c为c-myc基因蛋白免疫印迹试验产物电泳图。
从图8a和图8c可以看出,随着浓度增加聚酰胺pa1,pa3,pa5和pa6对应的c-mycmrna和蛋白表达的条带相对变暗,采用quantityone软件对图8a和图8c进行分析,结果分别见图8b和8d。
从图8a、图8b、图8c和图8d中的结果可知,在聚酰胺pa1~pa6存在下,与不加聚酰胺的对照组相比,a549细胞的c-myc基因的转录水平呈现不同程度的下调。通过进一步数据分析(图8b),聚酰胺pa1~pa6在1μm浓度时,对a549细胞中c-myc基因的转录并没有明显的抑制作用。但当聚酰胺的浓度达到10μm时,聚酰胺pa1,pa3,pa5和pa6对c-myc基因的转录的抑制作用明显增强,c-myc的mrna表达水平分别下降了:54.60%,44.95%,60.97%,63.81%。而聚酰胺pa2和pa4在所有浓度下对c-myc基因的转录的抑制作用都较弱。
聚酰胺pa1~pa6对a549细胞内c-myc蛋白水平的抑制(图8c和图8d)。根据westernblot实验结果显示,聚酰胺pa1~pa6对c-myc蛋白水平的影响情况与pcr的结果一致。在1μm浓度下,聚酰胺的抑制作用除pa5和pa6外不明显。在聚酰胺pa1~pa6的加药浓度为10μm时,a549细胞中c-myc蛋白水平分别下调了53.2%、35.7%、60.4%、30.0%、75.7%和85.6%。综合聚酰胺pa1~pa6对c-mycmrna和蛋白水平的影响,聚酰胺pa1,pa3,pa5和pa6对c-mycmrna和蛋白水平的抑制最明显,尤其,聚酰胺pa5和pa6,而聚酰胺pa2和pa4的作用较弱。
聚酰胺pa1~pa6对sgc7901细胞内c-myc表达的影响见图9a和图9b;从图9a和图9b中的结果可知,在聚酰胺pa1~pa6存在下,与不加聚酰胺的对照组相比,sgc7901细胞的c-myc基因的转录水平呈现不同程度的下调,且呈现浓度依赖的关系。低浓度时聚酰胺对sgc7901细胞中c-myc基因转录的抑制作用并不明显。通过进一步数据分析(图9b),聚酰胺pa2和pa4对sgc7901细胞中c-myc基因转录的抑制作用不明显。当加药浓度达到10μm时,聚酰胺pa1,pa3,pa5和pa6对c-myc基因转录的抑制作用明显增强,c-myc基因转录水平分别下降了40.9%,45.6%,74.8%和85.6%。
聚酰胺pa1~pa6对sgc7901细胞内c-myc蛋白水平抑制的影响见图9a和图9b。从图9c和图9d中可以看出,当加药浓度为5μm时,聚酰胺pa1,pa3,pa5和pa6对sgc7901细胞中c-myc蛋白的表达有明显的抑制作用,而且抑制作用呈现浓度依赖性。当浓度达到最大10μm时,聚酰胺pa1,pa3,pa5和pa6作用后的sgc7901细胞中的c-myc蛋白水平分别下降了72.2%、90.5%、90%和95.9%。聚酰胺pa2在最大加药浓度下才出现了抑制作用,而聚酰胺pa4在任何浓度下都没有明显的抑制作用。
通过rt-pcr实验和westernblot实验,探究聚酰胺pa1~pa6对a549细胞和sgc7901细胞中原癌基因c-myc转录和表达的影响情况。从图结果说明聚酰胺pa1~pa6对靶标细胞中c-myc基因表达有不同程度的抑制作用,且抑制作用基本呈现浓度依赖的特性。聚酰胺pa2和pa4在低浓度下对细胞c-mycmrna水平并没有明显的抑制效果,但当浓度增加到10μm时,聚酰胺pa2和pa4对细胞内的c-myc蛋白水平表达起到了微弱的抑制作用。而聚酰胺pa1,pa3,pa5和pa6在加药浓度达到5μm就能够明显地抑制c-myc基因的转录和表达。结果表明聚酰胺pa1,pa3,pa5和pa6对sgc7901细胞c-myc的表达有更好的抑制作用。
实施例9:聚酰胺对a549细胞和sgc7901肿瘤细胞迁移的影响
划痕实验材料与方法(三):
(1)a549,sgc7901肿瘤细胞的培养同实验材料与方法(一)。
(2)划痕实验的方法:待肿瘤细胞至对数生长期,接种于合适的细胞培养孔板,用枪头在孔板底部划直线,记录并记录划痕的位子,然后加入无血清的培养液和不同浓度的化合物,合适温度培养24小时,用倒置显微镜拍照记录肿瘤细胞向划痕区迁移的相对距离。实验结果如图10、图11、图12和图13所示。
其中在图10中,图片a~f分别为聚酰胺pa1~pa6对a549细胞迁移的抑制作用的图片。在图12中,图片a~f分别为聚酰胺pa1~pa6对sgc7901细胞迁移的抑制作用的图片。
通过细胞划痕实验结果表明,在0.1μm浓度下,聚酰胺pa1,pa3,pa5和pa6对a549细胞和sgc7901细胞的迁移有一定的抑制作用,当浓度为0.5μm时,抑制两种肿瘤细胞迁移的能力较明显,抑制率都在60%以上,其中聚酰胺pa6对sgc7901细胞的迁移抑制率达到了96.5%以上。
实施例10:聚酰胺对a549细胞和sgc7901肿瘤细胞凋亡和周期的影响
应用流式细胞术考查六种聚酰胺对c-myc基因高表达的a549和sgc7901两种肿瘤细胞的杀伤及抑制增殖的方式。
实验材料与方法(四)
材料:凋亡试剂盒(bd,美国)
培养基的组成:同实验材料与方法(一)
肿瘤细胞的培养与收集:同实验材料与方法(一)
实验材料与方法:
(1)a549,sgc7901肿瘤细胞的培养同实验材料与方法(一)。
(2)细胞凋亡的实验方法:将细胞(a549和sgc7901)细胞以每孔1×105个细胞接种在12孔板上,并在含有10%fbs的1ml1640补充物中生长。将细胞维持在37℃和5%co2。20小时后,更换培养基,向各孔中加入1ml不同浓度的聚酰胺。将细胞在37℃及5%co2下孵育24小时。然后弃去培养基。用pbs(含2%fbs)洗涤细胞。用不含edta的胰蛋白酶消化细胞,并收集在离心管中。用冷pbs离心并洗涤细胞。添加染料结合缓冲液,使最终细胞浓度为1×106个细胞/ml。将100μl细胞悬浮液转移至新的流式管。然后加入染料a。在室温下孵育15分钟,避光。加入染料b并孵育10分钟。最后添加400μl染料结合缓冲液。使用流式细胞术检测和记录625nm和488nm的7-aad荧光和pi荧光。所有组都有三组作为并行测试。
(3)检测细胞周期的方法:将细胞以每孔2×105个细胞接种在六孔板上,并在含有10%fbs的2ml细胞培养液补充物中生长。将细胞维持在37℃及5%co2。20小时后,通过使用不含fbs的培养基替换细胞培养液,对细胞进行饥饿。一夜后,用pbs冲洗培养基和碎片,并用含有不同浓度的化合物的新鲜细胞培养液代替。对照组由1%dmso表示。将细胞在37℃及5%co2的饱和湿度下培养36小时。接下来,收集细胞并用pbs洗涤两次。将细胞用合适浓度的乙醇(预冷)在4℃下固定过夜。通过离心除去乙醇并用pbs洗涤两次。将细胞转移到流式管中。加入rna酶后,将试管加入37℃水浴中水浴。最后,向每个管中加入周期染料,并在4℃在黑暗中染色25分钟。使用流式细胞术检测和记录488nm的细胞周期。(facscalibur,bd,usa)。所有组都有三组作为平行测试。
聚酰胺pa1~pa6分别加入,药物干预后的a549,sgc7901经过流式细胞仪检测,用modfit软件分析数据(结果见图14a、图14b、图15a和图15b),可以看出随着聚酰胺浓度的增加肿瘤细胞凋亡比例逐渐增加,表明聚酰胺能够通过诱导细胞凋亡的方式杀伤肿瘤细胞,其中pa5和pa6诱导凋亡的作用最为显著。
从图14a和图14b中可以看出,当加药浓度为6μm时,聚酰胺pa1,pa3,pa5和pa6对a549细胞具有明显的杀伤作用,而且抑制作用呈现浓度依赖性。当浓度达到最大12μm时,聚酰胺pa6作用后的a549细胞的总体凋亡率在60%以上。
从图15a和图15b中可以看出,当加药浓度为6μm时,聚酰胺pa1,pa3,pa5和pa6对sgc7901细胞具有明显的杀伤作用,与对照组相比,早期凋亡和晚期凋亡的比例均有增加,而且抑制作用呈现浓度依赖性。当浓度达到最大12μm时,聚酰胺pa5和pa6作用后的sgc7901细胞的总体凋亡率都在60%以上。聚酰胺pa2和pa4在最大加药浓度下也显示了一定的杀伤作用,不过作用效果较弱。而聚酰胺pa1和pa3的杀伤作用在两者之间。并且与a549细胞相比,聚酰胺的杀伤作用普遍偏强。
肿瘤细胞凋亡周期检测:聚酰胺pa1~pa6分别加入,药物干预后的a549,sgc7901经过流式细胞仪对药物干预后的肿瘤细胞进行细胞周期检测,用周期软件分析数据(结果见图16a、图16b、图17a和图17b),可以看出随着聚酰胺浓度的增加肿瘤细胞g1期比例逐渐增加,药物的加入使得肿瘤细胞阻滞在g1期,结果表明聚酰胺能够干预细胞周期,并通过将细胞周期阻滞在g1期的方式抑制肿瘤细胞的增殖。
细胞周期的实验结果显示,聚酰胺在3μm浓度下,对a549细胞的周期开始干预,与对照组相比,聚酰胺加入后,处于g1期的a549的细胞比例明显增加,表明周期阻滞在g1期。通过图16b可以看出,在聚酰胺pa1~pa6的加药浓度为6μm时,与对照组相比a549g1期的细胞比例增加30%以上。聚酰胺pa5和pa6g1增加的比例在40%左右,作用最为显著。
图17a和图17b的实验结果显示,聚酰胺在3μm浓度下,sgc7901细胞g1期比例增加,由图17a可以看出,当聚酰胺的浓度为6μm时s期和g2期的峰值很低,细胞基本集中在g1期。通过图16b可以看出,在聚酰胺pa1~pa6的加药浓度为6μm时,与对照组相比sgc7901g1期的细胞比例增加在40%左右。
本发明研究结果表明:聚酰胺作为c-myc基因表达抑制剂,聚酰胺pa1,pa3,pa5和pa6对a549细胞和sgc7901细胞的dna分子水平还是细胞水平上都有一定的抑制和杀伤作用,其中聚酰胺pa6为最佳的靶基因调控剂和靶细胞抑制剂。聚酰胺pa6不仅具有较强的抗肿瘤活性,而且具有较好的特异性,因此聚酰胺pa6具有潜力被开发成靶向c-myc基因的抗肿瘤药物。
本说明书所述的内容仅仅是对发明构思实现形式的列举,本发明的保护范围不应当被视为仅限于实施例所陈述的具体形式。
- 还没有人留言评论。精彩留言会获得点赞!