一种基于深度迁移学习的小分子药物虚拟筛选方法及其应用与流程
本发明涉及一种小分子药物筛选方法及其应用,尤其涉及一种基于深度迁移学习的小分子药物虚拟筛选方法及其应用。
背景技术:
小分子药物筛选往往需要通过高通量实验技术到数量巨大的化合物数据库中测定靶点与化合物作用的生物活性值,来筛选先导化合物。然而,实验的方法耗时耗力,更坏的是,往往可得到的化合物数量非常有限,而且并不是所有的药物靶点都适合于高通量筛选实验。因此,基于计算的虚拟筛选,它通过模拟目标靶点与候选药物之间的相互作用,在小分子药物设计中得到了广泛的应用。
基于计算的虚拟筛选可以分为两类,即基于受体的虚拟筛选和基于配体的虚拟筛选。基于受体的虚拟筛选通过模拟化合物与靶点的物理学相互作用来进行筛选,但需要知道靶点精确的三维结构,而且计算量很大。基于配体的虚拟筛选无需知道药物靶点三维结构,它利用已知活性的化合物,根据化合物的形状相似性或药效团模型在化合物数据库中进行搜索。
由于可用数据的急剧增加、各种化学描述符的产生和机器学习方法的巨大发展,配体虚拟筛选在小分子药物设计中得到了大量的应用。目前靶向已有靶点的小分子药物设计空间已经接近饱和,开发新药需要发现新的能够成药的靶点。然而,新的药物靶标往往已知活性配体样本信息不充分,已有的配体虚拟筛选方法的成功依赖于大量的数据样本,当已知活性配体样本信息不充分时,配体虚拟筛选难于得到好的预测性能。
技术实现要素:
发明目的:本发明提供了一种基于深度迁移学习的小分子药物虚拟筛选方法,用于解决小分子药物虚拟筛选中已知活性配体样本信息不充分而难于得到好用的虚拟筛选模型的问题。
技术方案:本发明提供一种基于深度迁移学习的小分子药物虚拟筛选方法,包括如下步骤:
s1、实验数据样本分为源域与目标域,先将源域作为输入,输入到基于配体的虚拟筛选的通用工具demo_new1中进行训练,经过收敛之后得到训练模型p=predictor(f,y);
s2、通过s1经过收敛之后得到的训练模型,得出权重矩阵w;
s3、将s1中的目标域中的实验数据样本作为输入,输入到我们的基于参数迁移的配体虚拟筛选的改进工具即demo_new2中;
s4、将通过s2得到的权重矩阵w输入到基于参数迁移的配体虚拟筛选的改进工具demo_new2中,作为目标域的初始化权重wi;
s5、参数迁移的配体虚拟筛选的改进工具demo_new2利用步骤s4得到的初始化权重wi和目标域中的实验数据样本进行使用fine-tune进行微调,继续训练直至收敛;
s6、在目标域中预测先导化合物与药物靶标相互作用的生物活性值,得到目标域加权分子指纹ft和预测值y,并输出最后预测结果的评价指标均方根误差rmse和相关系数r2。
s7、权重更新:用adam算法来对加权深度学习模型中的所有权重参数θ进行更新;
s8、判断模型优化是否达到预期的标准,如果没有,返回步骤2继续执行操作;反之,返回生成的新的目标域加权分子指纹ft。
s9、将由上述步骤之后源域达到最优模时的权重矩阵保存,作为目标域第一步的初始化权重,然后目标域重复以上步骤使用fine-tune进行微调,最终得到目标域的模型,即实现了借助源域的权重矩阵帮助目标域构建模型。
优选地,s1包括以下步骤:
s11、将源域作为输入,输入到基于配体的虚拟筛选的通用工具,即demo_new1中,初始化网络的参数,包括权重矩阵w和源域加权分子指纹fs;
s12、从给定一组用于训练的n个配体的数据d中随机抽取一批子集s;
s13、通过rdkit数据库,计算得到子集s中每一个分子的化学信息数据;
s14、提取需要的所有原子、近邻原子信息、化学键信息以及边信息;
s15、对分子进行累加操作依次得到每一模块单元的分子指纹fl;
s16、对s15中每一模块单元的分子指纹fl进行加权得到源域加权分子指纹fs;
s17、预测得到生物配体的活性:在本发明构建的用来预测生物配体的活性的回归预测模型如下式所示:
s18、计算目标损失函数:
优选地,s15中所述的每一模块单元的分子指纹f的生成由多个模块单元组成,每个模块单元包含四个层,依次为加和池化层,卷积层,卷积层,加和池化层。
进一步地,s15中所述的每一模块单元的分子指纹f包括原子及其相邻原子属性以及键类型;所述原子及其相邻原子的初始原子属性连接原子元素的单热编码,连接的氢原子数量和隐含的价数,以及芳香性指标;所述键类型为单键,双键,三键或者芳香键,键共轭或键成环和键的数量。
优选地,s15所述的每一模块单元的分子指纹f还包括相邻原子之间的区别信息,所述区别信息为在每个中心原子的相邻原子和中心原子之间的边信息。。
其中,上述边信息术语意思是一些可能有关联但是可能被忽略的信息:所述边信息是指在每个中心原子的相邻原子和中心原子之间的边缘上结合的特征,包括相邻原子的排列顺序信息等。
进一步地,所述的s6中预测先导化合物与药物靶标相互作用的生物活性值的步骤为:采用随机森林模型计算生物活性,随机森林是m个决策树的集合,具体包括:随机森林模型产生输出,其中是第m棵树的配体的预测值。
优选地,所述随机森林模型的训练程序如下:
(1)从n个配体的训练数据中抽取一个bootstrap样本数据集,通过随机抽样和训练数据集的替换生成n个训练样本,得到自举样本数据集;
(2)对于每个自举样本数据集,使用以下方案生成树:在每个节点上,选择随机的要素子集中的最佳分组,树生长到最大尺寸直到不再有可能分裂后,停止修剪;
(3)重复上述步骤,直到m个这样的树生长出来。
具体来说,上述步骤中,随机森林是m个决策树{t_1(x),…,t_m(x)}的集合,其中,x={x_1,…,x_k}是配体指纹的k维向量。随机森林模型产生m个输出结果{y^_1=t_1(x),…,y^_m=t_m(x)},其中,y^_m是第m个树的配体预测值。然后,再组合所有树的输出以产生一个最终预测y^,即为个体树预测的平均值。
给定一组用于训练的n个配体的数据d={(x_1,y_1),…,(x_n,y_n)},其中,x_i(i=1,…,n)是指纹向量,y_i是配体的生物活性值,训练程序如下:
从n个配体的训练数据中,绘制一个引导样本数据集,即通过从d中随机取样替代,产生n个训练样本;
对于每个引导样本数据集,使用以下方案生成树:在每个节点,选择随机获取到的特征子集中的最佳分割。树生长到最大尺寸,即直到没有更多的分裂是可能的,而不是进行修剪;
重复上述步骤直到m个这样的树生长出来。
本发明还提供一种基于深度迁移学习的小分子药物虚拟筛选方法的应用,所述应用为小分子药物虚拟筛选装置。
优选地,所述小分子药物虚拟筛选装置为基于配体的虚拟筛选的通用工具demo_new1、基于参数迁移的配体虚拟筛选的改进工具demo_new2、先导化合物与药物靶标作用的生物活性的预测工具demo_activity和先导化合物在药物靶标上的分子指纹的生成工具build_wdl_fp;
其中,所述基于配体的虚拟筛选的通用工具demo_new1提供自主开发药物靶标的虚拟筛选工具;输入:smiles格式的化合物及其生物活性值;输出:模型的性能参数,所述模型的性能参数为rmse和r2值;
所述基于参数迁移的配体虚拟筛选的改进工具demo_new2的输出:迁移所需的权重矩阵、fine-tune功能和模型性能参数,所述模型性能参数为rmse和r2值;输入:miles格式的化合物及其生物活性值;
所述demo_activity预测先导化合物与药物靶标相互作用的生物活性值,并将其应用于针对这些药物靶标的新药设计,药物副作用的预测及药物研发风险的评估,输入:smiles格式的化合物,输出:与这些gpcr药物靶标作用的生物活性值;
所述build_wdl_fp得到先导化合物在药物靶标上的多种短的分子指纹,用于化合物的相似性搜索、药效基团搜索等;输入:smiles格式的化合物;输出:分子指纹。
有益效果:本发明提供的小分子药物虚拟筛选方法及其应用能够在已知活性配体样本信息不充分的情况下仍得到有效的虚拟筛选模型,而不需要依赖大量的数据样本。
附图说明
图1为本发明方法的前馈结构图;
图2为本发明方法的步骤示意图;
图3为本发明应用中所述装置的结构示意图。
具体实施方式
我们以一组数据集输入作为例子进行介绍具体实施方式:
表1
如上表1所示,这是我们经过生物技术筛选后得到的数据集,它们因为都同属于同源蛋白所以被分为一组,有着共同的家族这里我们称之为groupa,其中a1-a6是我们的目标域,即我们所针对的小样本数据集,他们的数量在一百多到一千多不等,这对我们做深度学习十分不利,所以我们又找到我们的源域,即as1,as2,他们有几千个样本数量。我们要做的就是用源域来提高目标域的训练效果,具体实施步骤如下:
1.将源域作为输入,输入到我们的基于配体的虚拟筛选的通用工具demo_new1中进行训练:
(1)初始化网络的参数,包括权重矩阵w,分子指纹f0;
(2)从给定一组用于训练的n个配体的数据d中随机抽取一批子集s;
(3)通过rdkit数据库,计算得到子集s中每一个分子的化学信息数据;
(4)提取需要的所有原子、近邻原子信息、化学键信息以及边信息;
(5)对分子进行累加操作依次得到每一模块单元的分子指纹f,即分子指纹f是每一模块单元的累加;
(6)将每个模块单元得到的分子指纹f(记为fl)加权组合在一起,生成新的加权分子指纹fi。加权分子指纹生成部分只包含一层,即对由各模块单元生成的分子指纹进行加权得到新的加权分子指纹fi;
(7)预测得到生物配体的活性。由于药物靶标与配体相互作用的生物活性值跨度太大,在本发明构建的回归预测模型中,使用的活性值为-log10v,其中v是生物活性值,以此来缩小生物活性值的跨度。生物活性生成部分由两个全连接层构成。
(8)计算目标损失函数。通过使用训练数据集中配体分子个数、分子的真实活性值与预测活性值,以及加权深度学习模型中需要求解的权重参数,进行预测活性值的优化,来达到模型的最小化预测活性值与真实活性值的均方误差的目的。
2、通过s1经过收敛之后得到的训练模型,得出权重矩阵w;
3、将s1中的目标域中的实验数据样本作为输入,输入到我们的基于参数迁移的配体虚拟筛选的改进工具即demo_new2中;
4、将通过s2得到的权重矩阵w输入到基于参数迁移的配体虚拟筛选的改进工具demo_new2中,作为目标域的初始化权重wi;
5、参数迁移的配体虚拟筛选的改进工具demo_new2利用步骤s4得到的初始化权重wi和目标域中的实验数据样本进行使用fine-tune进行微调,继续训练直至收敛;
6、在目标域中预测先导化合物与药物靶标相互作用的生物活性值,得到目标域加权分子指纹ft和预测值y,并输出最后预测结果的评价指标均方根误差rmse和相关系数r2。
7、权重更新:用adam算法来对加权深度学习模型中的所有权重参数θ进行更新;
8、判断模型优化是否达到预期的标准,如果没有,返回步骤2继续执行操作;反之,返回生成的新的目标域加权分子指纹ft。
9、将由上述步骤之后源域达到最优模时的权重矩阵保存,作为目标域第一步的初始化权重,然后目标域重复以上步骤使用fine-tune进行微调,最终得到目标域的模型,即实现了借助源域的权重矩阵帮助目标域构建模型。
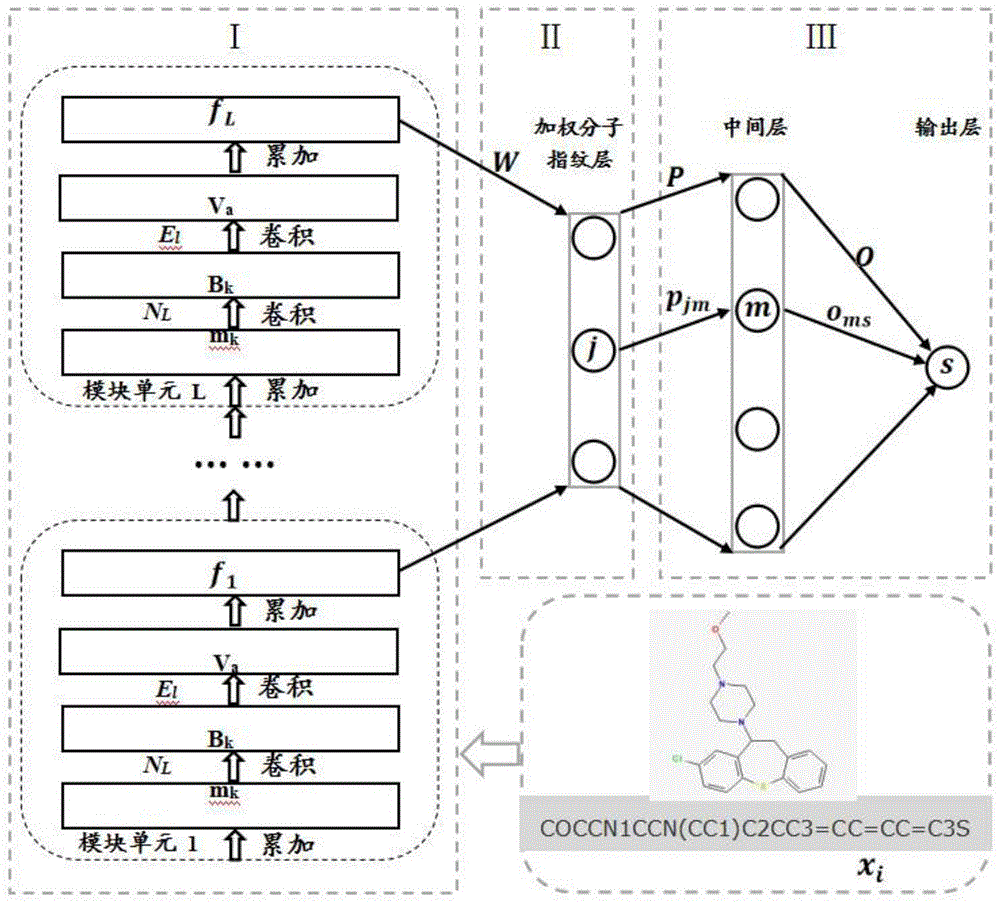
具体过程如图1所示,其中目标化合物如xi所示,本发明提出基于深度迁移学习的小分子药物虚拟筛选方法模型,即wdl-tl的前馈结构图,其包括三个部分,即基于模块单元的分子指纹生成(i)、加权分子指纹生成(ii)和生物活性生成(iii)。
基于模块单元的分子指纹的生成部分,包含了多个模块单元。对于每个模块单元,都划分成4层,分别为累加,卷积,卷积和累加这四种操作。
加权分子指纹生成部分只包含一层,即对由各模块单元生成的分子指纹进行加权得到新的分子指纹,即本发明所述的加权分子指纹。生物活性生成部分可以由两个全连接层构成。
起初,本发明给定配体分子数据集d={(x1,y1),...,(xn,yn)},其中i=1,...,n,其中xi表示第i个配体分子,yi表示其生物活性值。对配体分子xi,假设其含有ai个原子,本发明通过rdkit工具得到每个原子的属性向量mj,其中j=1,...,ai。
假设本发明的模型含有l个模块单元,对于第l个模块单元,假设配体分子xi中的任意原子α属性向量为ma,其含有na个近邻原子。
首先,本发明先通过累加操作考虑了原子α的所有近邻原子信息,及利用如下公式:
然后,再通过第一个卷积操作考虑了原子α的连接化学键的信息,利用如下公式:
其中,l∈[1,l];v代表原子α的连接化学键数量,v∈[1,5];
继而,本发明利用第二个卷积操作计算
ca=s(mahl)
其中,l∈[1,l];hl为连接权重向量,需要迭代更新;s(.)表示作为索引的可微分类比的softmax函数,即
最后,本发明再对分子进行累加操作依次得到每一模块单元的分子指纹f,即
f=f+ca
对分子xl,本发明将每个模块单元得到的分子指纹f(记为fl)加权组合在一起,生成新的加权分子指纹fi,利用公式
其中l是模块单元的数量,l∈[1,l],w是模块单元生成的分子指纹与加权分子指纹层之间的连接权重;σ(.)为relu激活函数,fi包括目标域加权分子指纹ft及源域加权分子指纹fs。
得到加权分子指纹fi后,本发明通过两个全连接层得到配体分子xi预测活性值,假设加权分子指纹层第j个神经元与中间层第m个神经元之间的连接权重为pjm,即
zm=σ(∑pjmfij)
假设中间层第m个神经元与输出层神经元s之间的连接权为oms,则
其中σ(.)为relu激活函数。
我们将部分实验的结果展示如下:如下表2所示,我们采用as1作为源域,将as1的权重矩阵赋给a1-a6的目标域,然后通过对比不同迭代轮数之后的训练结果,并于不采用迁移直接对a1-a6的样本进行本方法的测试结果做对比,即在权重初始化阶段仅采用随机初始化的结果,记作wdl-rf2,迁移后的结果记作tr-wdl-rf2,斜体加粗的数据为最佳数据,可见在迁移之后小样本数据集都有提升效果,并且有的效果很明显,说明我们方法的有效性数据与文字相对应来说明有效性。
表2。
- 还没有人留言评论。精彩留言会获得点赞!