一种基于非癌组织突变信息的癌症预测系统及其构建方法
1.本发明属于人工智能领域,具体涉及一种基于非癌组织突变信息的癌症预测系统及其构建方法。
背景技术:
2.尽管靶向癌症治疗和肿瘤免疫治疗已成功治愈了许多患者或显著提高了某些疾病的总体存活率,但癌症的检测和治疗仍然是一个需要面对的严重问题。及时有效地诊断癌症,从而促进早期患者的预干预和治疗,是提高疾病总体生存率的关键因素之一。目前用于早期癌症筛查的常规方法主要是影像学检测,但是存在射线剂量大和费用较高等原因,不适合经常使用。目前基于血液抗原的癌症检测,例如对前列腺特异性抗原(psa),肿瘤相关抗原(ca-125)和癌胚抗原(cea)的检测,这些检测只能针对单一或少量癌症类型,并且存在较高的假阳性情况。因此,临床癌症检测领域迫切需要新的血液检测方法以帮助医生进行早期诊断和筛查。
3.另一方面来说,由于遗传以及环境因素的影响,个体的基因组会发生基因突变,而其中某些“驱动”突变的产生加上突变的积累,最终可能会导致癌症的发生。因此,对个体基因组的突变检测有希望来预测癌症的发生。举例来说,brca1被一直认为是乳腺癌和卵巢癌的易感基因,该基因发生突变会增加患癌症的风险,但在所有乳腺癌女性中,只有约3%-8%的患者携带brca1或brca2突变。同样,brca1突变仅在约18%的卵巢癌中可见。与此同时,随着下一代高通量测序技术的迅速发展,一些大型的合作项目,如千人基因组计划(1000genomes project)和癌症基因组图谱计划(tcga,the cancer genome atlas)得以开展,提供了丰富的病人和正常人的基因组信息,为利用癌症的基因组变异进行癌症预测提供可能。虽然泛癌分析发现了一些基因,如tp53和pik3ca,这些基因与大多数癌症中超过10%的患者高度相关,但最新研究发现,这些癌症驱动基因的突变,同样在正常个体的血液和组织中普遍存在,这提示我们仅凭单个基因的突变来预测癌症风险是不可行的。因此,人们开始利用多个关键位点的突变来进行癌症预测。目前已有的方法主要为利用支持向量机(svm)对乳腺癌和多发性骨髓瘤进行预测,但预测准确率都仅为70%左右,并且这些研究所用的样本仅为百例左右,结果并不足以证明该方法的可靠性。所以,癌症筛查的领域迫切需要更为先进和强大的方法,可以利用个体基因组上的大量突变来进行癌症风险预测,并且需要大量的测试集来证明方法的可靠性。
4.最近几年,大量的深度学习研究极大地促进了人工智能技术的发展,并已广泛应用于精准医学领域,包括药物分子设计,医学影像诊断,疾病驱动基因/突变预测等。但其在癌症诊断领域的应用主要是基于大量临床图像的分析,还没有研究证明深度学习在癌症的早期诊断能够发挥很大的作用。
技术实现要素:
5.有鉴于此,本发明利用深度学习方法,构建深度神经网络模型来学习已有数据库
中大量的癌症病人与正常人的基因组变异信息,并且能够对血液来源的样本进行癌症风险预测,从而建立一个能应用于癌症早期预测的系统。
6.一方面,本发明提出一种基于非癌组织突变信息的癌症预测系统,所述癌症预测系统包括顺序连接的一个输入层、多个隐藏层和一个输出层;
7.其中所述输入层用于输入突变信息,所述突变信息为将外显子切分成的预定长度的窗口;
8.各隐藏层均包含m个全连接层,每个全连接层有n个节点,m和n为大于1的正整数,各隐藏层之间嵌入有maxout激活函数,其中,全连接层中的每个节点均进行如下线性变换:
9.z
ij
=x
t
w
ij
+b
ij
10.其中,z
ij
表示第i个神经元的第j个激活单元,x
t
表示输入的转置,w
ij
和b
ij
是系统需要学习的参数,分别代表输入层到激活单元z
ij
的权重矩阵和偏置向量;所述maxout激活函数对线性变换所得节点信息进行非线性变换得到n个节点,并输出到顺序连接的下一层,其中,maxout激活函数的输出是选择激活单元中的最大值:
[0011][0012]
其中,k代表maxout神经元激活单元组中激活单元的个数,x代表激活函数的输入;
[0013]
所述输出层接收由最后一个隐藏层得到的n个节点,并输出两个节点,分别表示预测个体患癌症的概率或正常的概率。
[0014]
在一些实施例中,在每相连两个隐藏层中加入dropout对前个隐藏层的输出进行正则化,避免maxout神经元可能带来的过拟合现象;
[0015]
r~bernoulli(p)
[0016][0017]
其中,p代表断开神经元连接的比率,伯努利函数bernoulli随机生成包含0,1的向量,r为服从伯努利分布的向量,h为输入向量,为随机断开连接后的输出向量。
[0018]
在一些实施例中,所述输出层输出的两个节点利用softmax激活函数进行归一化处理。在一些实施例中,所述归一化处理的方法为:
[0019][0020]
其中,σ表示激活函数,z表示由z1,...,z
k
组成的向量,k表示输入的节点数,z
j
表示第j个节点的输入,e为自然常数。
[0021]
另一方面,本发明提出一种所述癌症预测系统的构建方法,包括:
[0022]
获取癌症患者的突变信息和正常人的遗产变异信息,并将其中所有癌症患者的突变信息和一部分正常人的遗产变异信息作为训练集;
[0023]
将每个外显子切分成预定长度的窗口,保留至少在两个癌症患者中存在突变的窗口;
[0024]
将癌症患者和正常人的突变信息转变为窗口
×
样本的二值化矩阵,从所述训练集中随机抽取与癌症患者数量相同的二值化矩阵,并分别与癌症患者的窗口矩阵加和并二值化作为训练集,其中抽取的二值化矩阵互不重叠;
[0025]
构建包括顺序连接的一个输入层、多个隐藏层和一个输出层的所述癌症预测系
统;利用训练集训练系统,在首次收敛的区间中选择准确率最高的系统为最终的癌症预测系统。
[0026]
在一些实施例中,所述窗口的长度为50-300bp(例如100bp、150bp、200bp、250bp等)。
[0027]
在一些实施例中,所述方法还包括对所述癌症预测系统进行评估的步骤。
[0028]
在一些实施例中,通过绘制受试者操作特性(roc)曲线和精准率-召回率(precision-recall,pr)曲线来评估所述癌症预测系统的分类性能。
[0029]
在一些实施例中,评估指标选自测试集准确率(accuracy)、敏感性(sensitivity)、特异性(specificity)、roc曲线下的面积(auc)、平均精度(ap)中的一种或多种。
[0030]
又一方面,本发明还提出一种基于非癌组织突变信息的癌症预测装置,包括顺序连接的输入模块、数据处理模块和输出模块;
[0031]
其中所述输入模块用于输入突变信息,所述突变信息为将外显子切分成的预定长度的窗口;
[0032]
所述数据处理模块包括多个隐藏层,各隐藏层均包含m个全连接层,每个全连接层有n个节点,m和n为大于1的正整数,各隐藏层之间嵌入有maxout激活函数,其中,全连接层中的每个节点均进行如下线性变换:
[0033]
z
ij
=x
t
w
ij
+b
ij
[0034]
其中,z
ij
表示第i个神经元的第j个激活单元,x
t
表示输入的转置,w
ij
和b
ij
是系统需要学习的参数,分别代表输入层到激活单元z
ij
的权重矩阵和偏置向量;所述maxout激活函数对线性变换所得节点信息进行非线性变换得到n个节点,并输出到顺序连接的下一层,其中,maxout激活函数的输出是选择激活单元中的最大值:
[0035][0036]
其中,k代表maxout神经元激活单元组中激活单元的个数,x代表激活函数的输入;
[0037]
所述输出模块接收由所述数据处理模块的最后一个隐藏层得到的n个节点,并输出两个节点,分别表示预测个体患癌症的概率或正常的概率。
[0038]
在一些实施例中,在所述数据处理模块中,在每相连两个隐藏层中加入dropout对前个隐藏层的输出进行正则化,避免maxout神经元可能带来的过拟合现象;
[0039]
r~bernoulli(p)
[0040][0041]
其中,p代表断开神经元连接的比率,伯努利函数bernoulli随机生成包含0,1的向量,r为服从伯努利分布的向量,h为输入向量,为随机断开连接后的输出向量。
[0042]
在一些实施例中,所述输出模块输出的两个节点利用softmax激活函数进行归一化处理。
[0043]
在一些实施例中,所述归一化处理的方法为:
[0044][0045]
其中,σ表示激活函数,z表示由z1,...,z
k
组成的向量,k表示输入的节点数,z
j
表示
al.,whole exome sequencing suggests much of non-brca1/brca2 familial breast cancer is due to moderate and low penetrance susceptibility alleles.plos one,2013)。
[0065]
本发明以乳腺癌为例来评估癌症预测系统能否利用全基因组变异来进行癌症预测,该癌症的突变状态在基于突变基因的无监督聚类中显示出广泛的异质性,因此乳腺癌是一个很好的例子。已有的研究利用具有高体细胞突变的基因作为癌症分类的特征,结果并不好,这表明广谱的突变图不能直接用于分类或预测癌症。本发明在分析中使用窗口策略,有效的降低了特征数。本发明收集了大量独立的wes病人数据集,并与其他机器学习方法作比较,以评估深度学习模型的准确性和稳健性。需要说明的是,本发明的癌症预测系统并不仅适用于乳腺癌的预测,还可以适用于其它各种癌症,只要有足够的样本数据即可。本发明将该系统命名为miscan(maxout inferred snv-based cancer prediction model)。
[0066]
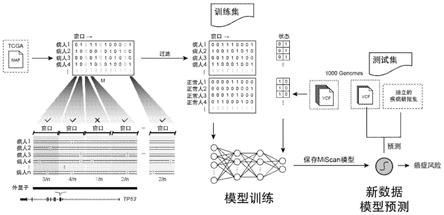
如图1所示,miscan模型具体的构建以及评估过程如下:
[0067]
一、miscan模型的构建
[0068]
1、下载tcga中包含986个乳腺癌病人的体细胞突变信息(文件格式为maf)以及千人基因组中第三阶段的2,504个正常人的遗产变异信息(文件格式为vcf)。将正常人数据集随机切分出80%(n=2,003)的数据作为训练集,剩余的作为测试集。
[0069]
2、将每个外显子切分成100bp长的窗口,每个外显子末端余下的窗口长度可能小于100bp。接下来进行窗口的过滤,如果至少有两个病人在某个窗口中存在突变(可以不是同一突变),则该窗口被保留下来,否则该窗口则被过滤掉。最后有13,885个有效窗口被保留下来。
[0070]
3、将病人和正常人的突变信息转变为窗口
×
样本的二值化矩阵。每一行是一个窗口特征,每一列是一个样本,如果某个样本在某个窗口中存在突变,对应的值为1,否则就为0。为了平衡训练集中的病人和正常人的数量,同时利用模型发现复杂的突变相关网络,从训练集的正常人中随机抽取两个互不重叠的、包含986个人信息的二值化矩阵,并分别与病人的窗口矩阵加和、二值化。最终的训练集数据为13,885个窗口特征和3975个样本组成的二值化窗口矩阵。
[0071]
4、使用python的keras api,以tensorflow作为后端来构建miscan模型。如图2所示,该模型包含一个输入层,七个隐藏层和一个输出层。每个隐藏层分别由32个全连接层组成组成;每个全连接层有128个节点,在隐藏层之间嵌入maxout激活函数以防止深度神经网络发生梯度消失或过度拟合。输出层以softmax作为激活函数。此外,在每个相邻层之间插入一个dropout(随机失活)层,并设置断开链接的比例为0.25(p=0.25)来防止过拟合。adam为模型的优化器(optimizer=adam),学习率设为0.001(lr=0.001);模型的损失函数为交叉熵损失函数(loss=
′
categorical_crossentropy
′
),评估指标为准确率(metrics=
′
accuracy
′
)。
[0072]
需要说明的是,模型中隐藏层的个数、全连接层的个数以及每个全连接层中的节点数由窗口特征、样本数量等因素决定,可根据实际情况适当选择。
[0073]
所构建的基于非癌组织突变信息的癌症预测系统从左到右依次为输入层i,隐藏层h1-h7,输出层o。具体构建步骤如下:
[0074]
第1步,将训练数据集嵌入到包含13885个节点的输入层i,得到模型第1层;
cancer is due to moderate and low penetrance susceptibility alleles.plos one,2013),经过数据预处理得到包含突变信息的vcf文件。
[0092]
2、将上述下载的病人数据和之前千人基因组剩余501个正常人(20%)的数据一起,作为测试数据集。
[0093]
3、将vcf文件转化为窗口
×
样本的二值化矩阵,1表示该样本在该窗口中存在变异。
[0094]
4、利用miscan对上述测试集进行预测,绘制受试者操作特性(roc)曲线和精准率-召回率(precision-recall,pr)曲线来评估miscan模型的分类性能。对每种方法根据预测值和真实标签计算相关指标——测试集准确率(accuracy),敏感性(sensitivity),特异性(specificity),roc曲线下的面积(auc),平均精度(ap)。
[0095]
5、对测试集数据进行下采样,以直接从vcf文件格式抽取一定比例的突变信息和直接从原始测序数据抽取一定的数据量两种方式来测试miscan模型的鲁棒性。
[0096]
6、为了评估基因对模型预测的影响,分别将每个基因对应的所有窗口去掉以后,重新进行训练和测试,并使用上述一样的条件来选取最优的模型,以确定模型的性能是否受到影响。
[0097]
本发明还提出一种基于非癌组织突变信息的癌症预测装置,包括顺序连接的输入模块、数据处理模块和输出模块;
[0098]
其中所述输入模块用于输入突变信息,所述突变信息为将外显子切分成的预定长度的窗口;
[0099]
所述数据处理模块包括多个隐藏层,各隐藏层均包含m个全连接层,每个全连接层有n个节点,m和n为大于1的正整数,各隐藏层之间嵌入有maxout激活函数,其中,全连接层中的每个节点均进行如下线性变换:
[0100]
z
ij
=x
t
w
ij
+b
ij
[0101]
其中,z
ij
表示第i个神经元的第j个激活单元,x
t
表示输入的转置,w
ij
和b
ij
是系统需要学习的参数,分别代表输入层到激活单元z
ij
的权重矩阵和偏置向量;所述maxout激活函数对线性变换所得节点信息进行非线性变换得到n个节点,并输出到顺序连接的下一层,其中,maxout激活函数的输出是选择激活单元中的最大值:
[0102][0103]
其中,k代表maxout神经元激活单元组中激活单元的个数,x代表激活函数的输入;
[0104]
所述输出模块接收由所述数据处理模块的最后一个隐藏层得到的n个节点,并输出两个节点,分别表示预测个体患癌症的概率或正常的概率。
[0105]
在所述数据处理模块中,在每相连两个隐藏层中加入dropout对前个隐藏层的输出进行正则化,避免maxout神经元可能带来的过拟合现象;
[0106]
r~bernoulli(p)
[0107][0108]
其中,p代表断开神经元连接的比率,伯努利函数bernoulli随机生成包含0,1的向量,r为服从伯努利分布的向量,h为输入向量,为随机断开连接后的输出向量。
[0109]
所述输出模块输出的两个节点利用softmax激活函数进行归一化处理。
[0110]
优选地,所述归一化处理的方法为:
[0111][0112]
其中,σ表示激活函数,z表示由z1,...,z
k
组成的向量,k表示输入的节点数,z
j
表示第j个节点的输入,e为自然常数。
[0113]
本发明中各功能模块可以是硬件,比如该硬件可以是电路,包括数字电路,模拟电路等等。硬件结构的物理实现包括但不局限于物理器件,物理器件包括但不局限于晶体管,忆阻器等等。所述计算装置中的计算模块可以是任何适当的硬件处理器,比如cpu、gpu、fpga、dsp和asic等等。所述存储单元可以是任何适当的磁存储介质或者磁光存储介质,比如rram,dram,sram,edram,hbm,hmc等等。
[0114]
所属领域的技术人员可以清楚地了解到,为描述的方便和简洁,仅以上述各功能模块的划分进行举例说明,实际应用中,可以根据需要而将上述功能分配由不同的功能模块完成,即将装置的内部结构划分成不同的功能模块,以完成以上描述的全部或者部分功能。
[0115]
实施例1
[0116]
一、样本信息
[0117]
从网上下载了已发表研究的乳腺癌全外显子数据,包括152例外周血的样本和360例癌旁组织的正常样本(数据来源为icgc数据库和两项研究:rheinbay,esther,et al.,recurrent and functional regulatory mutations in breast cancer.nature,2017;gracia-aznarez,francisco javier,et al.,whole exome sequencing suggests much of non-brca1/brca2 familial breast cancer is due to moderate and low penetrance susceptibility alleles.plos one,2013)。
[0118]
二、操作步骤
[0119]
1、数据预处理及预测
[0120]
(1)利用bwa软件来将原始的wes-seq fastq测序数据比对到参考基因组hg19。
[0121]
(2)使用picard来进行duplicate标记和基本质量分数重新校准(bqsr)。
[0122]
(3)使用gatk来寻找snv以及插入缺失(indel)
[0123]
(4)对寻找到的snv进行过滤。具体过滤参数设置如下:
[0124]
qd<2.0||fs>60.0||mq<40.0||mqranksum<-12.5||readposranksum<-8.0||sor>4.0
[0125]
其中,qd(qualbydepth)表示变异位点可信度除以未过滤的非参考read数;fs(fisherstrand)表示fisher精确检验评估当前变异是链偏差的可能性;mq(rmsmappingquality)为所有样本中比对质量的平方根;mqranksum表示根据参考和变异的比对质量来评估可信度;readposranksumtest通过变异在read的位置来评估变异可信度,通常在read的两端的错误率比较高;sqr(strandoddsratio)综合评估链偏差的可能性。
[0126]
(5)对寻找到的indel进行过滤。具体过滤参数设置如下:
[0127]
qd<2.0||fs>200.0||sor>10.0||mqranksum<-12.5||readposranksum<-8.0
[0128]
(6)将过滤后的snp和indel文件整合成最终的vcf文件格式。
[0129]
(7)将vcf文件转化为窗口
×
样本的矩阵。
[0130]
(8)将上述的病人数据(n=512)和千人基因组剩余样本的数据(n=501)合并到一起,作为完整的测试集。
[0131]
(9)读入已训练好的miscan模型,对测试数据进行预测,给出每个样本的预测概率值。
[0132]
2、性能测试
[0133]
主要从准确率,敏感性,特异性,roc曲线和pr曲线等方面对miscan和其他机器学习的方法进行测试和比较。
[0134]
3、鲁棒性检验
[0135]
(1)基于snv抽取的检验:从测试集最后得到的vcf文件中随机抽取10%~90%(每次间隔10%)的数据,然后进行数据处理和预测。该随机过程重复100次。
[0136]
(2)基于不同测序深度的检验:从突变发现前的原始文件中随机抽取10%~90%(每次间隔10%)的数据,然后进行数据处理和预测。该随机过程对每个测试集中的病人样本重复10次。
[0137]
4、基因重要性评估
[0138]
(1)去掉某个基因对应的所有窗口,对窗口削减后的训练集重新用相同的模型进行训练和测试。
[0139]
(2)重复(1)过程,直到所有的基因都被处理过。
[0140]
(3)分别以准确性,敏感性,特异性,auc,ap五个指标对每个基因删除后的测试效果进行升序排列。
[0141]
(4)选取十个在乳腺癌病人高频突变的驱动基因和2个导致乳腺癌遗传易感性的基因(brcal和brca2),并在图中标出它们的相对位置。
[0142]
三、结果总结
[0143]
1、模型构建与训练
[0144]
从图3能够看出,模型在训练过程中逐步收敛。在epoch(训练时期)达到100之后,模型的训练趋于稳定。将首次出现连续5个epoch的训练准确率大于99%,并且准确率偏差小于0.02的区段称为稳定区段(两条虚线之间的区段)。在该稳定区段中选出了训练准确率最高的模型(epoch=113)作为最终的miscan模型。
[0145]
2、模型性能评估
[0146]
为了评估模型的性能,将miscan与其他流行的机器学习方法进行比较,包括单个模型——决策树(dt),k近邻(knn),支持向量机(svm)以及两种集合方法——随机森林(rf)和梯度上升决策树(gbdt)。miscan具有最高的准确率——97%,而其他机器学习方法的预测准确率为86%(svm),73%(dt),63%(rf),56%(gbdt)和49%(knn)(图4,左)。
[0147]
此外,miscan对患者和正常个体的预测分别具有高灵敏度(100%)和高特异性(95%),优于其他方法,而一些方法如knn和gbdt错误地将大多数正常人都预测为病人,这显示了只有miscan的预测效果最佳,且没有任何预测偏好(图4,右)。
[0148]
同时从roc和pr曲线能够看出,miscan模型具有最强的分类性能,其auc和ap值分别达到了0.994和0.989,显著优于剩余的其他方法(图5)。
[0149]
进一步的,使用训练和测试数据集分析了miscan模型的预测概率分布(图6)。图中能够很明显的看出,miscan模型对于训练集的拟合较好,而且对测试集中病人和正常人的
预测概率值分别集中在0和1附近,说明miscan模型能够很明显的区分病人和正常人。对于所有测试样本预测结果的所有评估指标都展示在表1中。
[0150]
上述结果说明了相比于其他机器学习方法,miscan方法具有最优的分类性能和预测能力,对病人和正常人的预测不存在偏好性。
[0151]
表1:miscan和其他方法预测性能统计表
[0152][0153]
3、鲁棒性检验
[0154]
鲁棒性检验的目的有两个,一是在于评估方法的抗干扰性,二是评估方法对低质量数据的预测效果,从而检测该方法能否在将来以可观的成本应用于癌症的早期筛查。本发明首先对测试集中每个样本的vcf文件进行snv下采样。发明人注意到即使在数据覆盖的突变数目低至原始数据的10%时,miscan依旧具有极高的分辨能力——其auc和ap值均高于0.99,而原本分类性能较好的svm和rf随着数据覆盖snv数的下降,效果变得越来越差,这充分表明了miscan模型具有极强的稳定性(图7,上)。不同比例的snv输入下,miscan在预测准确率上也显示了最好的稳定性(图7,下)。
[0155]
由于测序深度是影响snv检测的关键因素之一,本发明也检查这些方法在低测序深度下的表现。对测试集中的原始测序数据进行了下采样,这样可以更为真实地模拟低质量数据。发明人注意到即使测序的数据量低至一百万个读长(1m),miscan也能够以高灵敏度识别这些患者。相比之下,随着测序深度的减少,其他机器学习方法的性能急剧下降,证实了miscan对这些模型的稳健性(图8)。由于外周血的基因组变异信息非常容易获得,因此miscan的高准确性和鲁棒性很适合于乳腺癌的早期诊断。
[0156]
3、基因重要性评估
[0157]
深度学习虽然具有强大的能力,但是其内部的黑箱性质使得研究者难以研究内部特征之间的关联。而在maxout模型中,评估每个基因对于模型的贡献显得尤为重要。为了评估每种基因变异对疾病识别的贡献,本发明设计了一种评估基因权重的算法,基本的原理是:对于一个特定的基因,去掉该基因对应的所有突变后重新进行模型的训练和预测(记为缺陷模型)。然后,根据不同的指标对所有缺陷模型进行排序,以说明每个基因的影响。
[0158]
令人惊讶的是,尽管某些乳腺癌相关基因(如pik3ca,map3k1,brca1等)的预测准确性的贡献可能比常规基因更强,但忽略任何单一基因并未显著降低分类和预测能力miscan(图9)。更有趣的是,任何单个基因的删除最多将预测准确度从99%降低到91%,并且分类指标(auc和ap)总是高于0.99,表明少数驱动基因的突变可能不会增加癌症的可能性。相反,许多热点中突变的积累和协同作用更为关键。这也可能反映了癌症发生和发展的异质性和复杂性。
[0159]
以上所述的具体实施例,对本发明的目的、技术方案和有益效果进行了进一步详
细说明,应理解的是,以上所述仅为本发明的具体实施例而已,并不用于限制本发明,凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1