一种治疗男性勃起功能障碍药物的制备方法与流程
本发明属于药物制剂领域,更具体地,涉及一种治疗男性勃起功能障碍药物的制备方法及片剂。
背景技术:
勃起功能障碍是一种常见疾病,成人男性中的约10%患有该疾病,据报道,目前全世界范围内约有1亿5千万名患有该疾病,美国有3千万名,韩国有250-300万名。由于社会的老龄化所导致的老年人口的增加、以及社会的产业化和急剧变化所导致的压力和过劳,因此,勃起障碍患者人数呈急剧增加的趋势。预计到2020年,患者人数将达到3亿以上,比目前的人数会增加两倍以上。
他达拉非(tadalafil)是一种可逆的、选择性pde5抑制剂,由礼来公司开发治疗男性勃起功能障碍(ed),2003年11月23日经fda批准上市,化学名为(6r-12ar)-6-(1,3-苯并二恶茂-5-基)-2-甲基-2,3,6,7,12,12a-六氢化吡嗪并[1',2'-1,6]-吡啶并[3,4-b]吲哚-1,4-二酮,结构如下:
他达拉非是几乎不溶于水并且极微溶于乙醇的结晶固体,为属于生物药剂学分类系统(biopharmaceuticsclassificationsystem,bcs)ii型或iv型的溶解性非常低的药物,已知他达拉非实质上为水不溶性物质,仅微量溶解于诸如甲醇、乙醇及丙酮等的部分有机溶剂中,生物利用度比较低。
在单次口服剂量用药以后,所观察到的他达拉非的血浆浓度最大值(cmax)在30分钟与6小时(2小时的中值时间)之间实现,但生物利用度尚未明确,故需研制药物制剂及制备方法,使生物利用度与原研药品一致并等效,同时提高稳定性,对于他达拉非在临床上广泛应用于治疗男性性功能障碍尤为重要。
技术实现要素:
本发明的目的在于克服现有技术,提供一种治疗男性勃起功能障碍药物的制备方法。该制备方法包括了特定的微粉化步骤,提高粉碎后的含他达拉非的粉末组合物的渗透性,同时以该粉末组合物制备的他达拉非片剂生物利用度与原研药品一致并等效,并且具有好的稳定性。同时,本发明的制备方法,特别适合工业化大规模生产。
本发明的上述目的是通过以下方案予以实现的:
一种治疗男性勃起功能障碍药物的制备方法,包括微粉化、辅料预处理、预混合、制粒、整粒、干燥、总混、压片步骤,所述微粉化为将他达拉非原料与部分甘露醇和/或预胶化淀粉混合后,过筛,进行气流粉碎处理,得到含他达拉非的粉末组合物,完成微粉化步骤;
他达拉非原料与部分甘露醇和/或预胶化淀粉的质量比为1:1~1:4。
甘露醇或者预胶化淀粉是制备片剂过程中的常用辅料,通常在制粒的步骤加入。而发明人通过大量试验总结分析发现,用适量的甘露醇或者预胶化淀粉与他达拉非原料混合,能使粉碎后的他达拉非粉末组合物具有更好的渗透性,以该粉末组合物制备的他达拉非片剂的生物利用度与原研药品一致并等效,并且具有好的稳定性。
优选地,他达拉非原料与部分甘露醇或预胶化淀粉的质量比为1:1~1:2。
一般地,微粉化处理的颗粒,已经达到足够小的尺寸,优选地,本发明中,微粉化后的颗粒的粒度为d90≤10um。
优选地,所述气流粉碎处理的条件为:进料气压0.2~0.6mpa,粉碎气压0.2~0.8mpa。在这种条件下,能更容易地达到本发明所述粒度,同时使他达拉非粉末组合物分散均匀。
优选地,所述气流粉碎处理的次数为1次。
优选地,所述过筛为过60目筛。
优选地,所述辅料预处理为将粘合剂溶解,并加入表面活性剂,搅拌至溶清,得到混合溶液。
更具体地,所述辅料预处理可以按照如下进行:将粘合剂加入热的纯化水,搅拌溶解,然后加入表面活性剂,搅拌均匀后,再加入常温纯化水,搅拌至溶清。
优选地,所述粘合剂为羟丙基纤维素,表面活性剂为十二烷基硫酸钠。
优选地,所述预混合为将他达拉非粉末组合物、甘露醇或者预胶化淀粉、低取代羟丙基纤维素、交联羧甲基纤维素钠混合均匀。
优选地,所述预混合在制粒机中进行。预混合的条件优选为在搅拌转速为2~5rps下,混合3~6min。
优选地,所述制粒为往预混合得到的混合物中喷雾加入预处理后的辅料(即所述混合溶剂),进行湿法制粒。
优选地,混合溶剂加入时,控制搅拌转速为1~6rps,切刀转速为6~10rps。
湿法制粒形成的软材,会进行过筛整粒,优选采用直径为1.0mm的筛进行整粒。
整粒后的颗粒的干燥,优选在流化床中进行。干燥的条件优选为进风温度50~70℃,进行80~100m3/h,干燥至lod≤3.0%。
在获得干燥的颗粒后,会将颗粒与润滑剂进行总混,为压片进行准备。
所述润滑剂可以是硬脂酸镁。
优选地,压片过程根据颗粒含量计算应压片重,硬度控制在30~80n。
优选地,压片后还包括包衣步骤,包衣增重质量控制在2~5%。
优选地,所述制备方法每次制备的片数为0.1~100万片。
基于本发明所述制备方法,本发明提供一种治疗男性勃起功能障碍药物他达拉非片剂,
包含以下重量计算组成成分,以生产1000片计算:
其中,
余下量的甘露醇与含他达拉非的粉末组合物中所含甘露醇使药物中的甘露醇总量为160g;
余下量的预胶化淀粉与含他达拉非的粉末组合物制备中所含预胶化淀粉使药物中的预胶化淀粉总量为140g。
与现有技术相比,本发明具有如下有益效果:
本发明通过对现有工艺的改变,以部分甘露醇和/或预胶化淀粉与他达拉非原料混合粉碎,得到他达拉非粉末组合物,该他达拉非粉末组合物的渗透性更好,以其制成的他达拉非片剂的生物利用度与原研药品一致并等效,同时具有较好的稳定性,对于他达拉非在临床上广泛应用于治疗男性性功能障碍尤为重要。同时,本发明的制备方法,特别适合工业化大规模生产。
附图说明
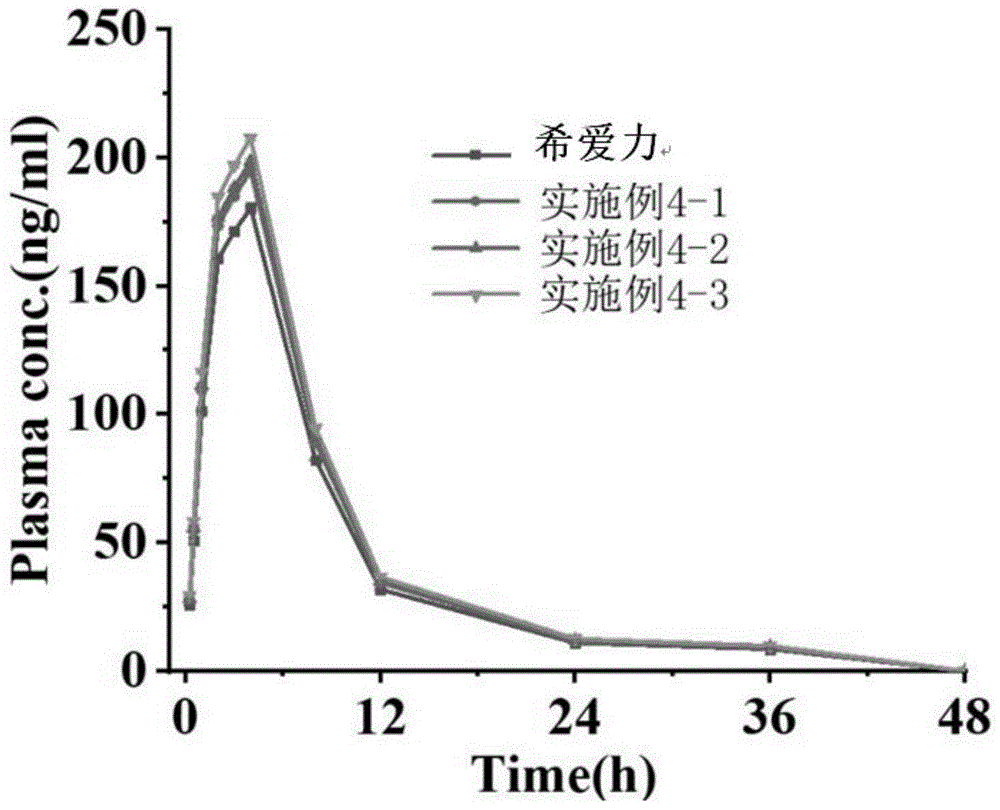
图1:实施例4与对照产品(希爱力)血药浓度曲线图。
具体实施方式
下面结合具体实施例进一步说明本发明的内容,但不应理解为对本发明的限制。在不背离本发明精神和实质的情况下,对本发明方法、步骤或条件所作的简单修改或替换,均属于本发明的范围;若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
预胶化淀粉购自湖州展望药业股份有限公司。
制备的他达拉非的微粉化粉末组合物,采用马尔文粒径检测仪进行粒径检测,并通过渗透性实验检测渗透性,以及后续按照工艺制备片剂的溶出度、有关物质、血浆浓度等检测数据反应制剂的生物利用度、稳定性。
实施例1
依照表1的处方制备1000片他达拉非片。
表1(1000单片用量及成分占比,各成分的计量单位为克
制备工艺如下:
微粉化:
他达拉非、甘露醇和/或预胶化淀粉按照表1中他达拉非粉末组合物的质量比混合过60目筛,经气流粉碎处理1次,形成他达拉非的粉末组合物。气流粉碎的条件为:进料气压0.4mpa,粉碎气压0.6mpa,采用粒径检测仪对他达拉非粉末组合物进行检测。
辅料预处理:
先将羟丙纤维素加入热纯化水,搅拌均匀,加入十二烷基硫酸钠,搅拌均匀后再加入常温纯化水,搅拌至溶清,得到混合溶液,待用。
预混合、制粒、整粒、干燥:
将他达拉非粉末组合物、剩余甘露醇、剩余的预胶化淀粉、低取代羟丙基纤维素、交联羧甲基纤维素钠加入到湿法混合制粒机中,搅拌转速3rps,混合5min,混合均匀。喷雾加入上述的羟丙纤维素和十二烷基硫酸钠混合溶液,搅拌转速4rps,切刀转速8rps,制软材,控制气压0.3mpa,喷完继续搅拌3min。将软材过筛整粒,用直径1.0mm筛整粒,之后将湿颗粒进行干燥,控制进风温度60℃,100m3/h,干燥至lod≤3.0%。
总混、压片:
将干颗粒过24目筛,将颗粒、硬脂酸镁加入混合机中,转速10rpm,混合8min。根据颗粒含量计算应压片重,硬度80n,压片,得到素片。
包衣:
将素片包衣,包衣增重控制2~5%。
实施例2
处方与实施例1相同,区别在于,微粉化过程中,气流粉碎的条件为:进料气压0.6mpa,粉碎气压0.2mpa,采用粒径检测仪对他达拉非粉末组合物进行检测。
实施例3
处方与实施例1相同,区别在于,预混合、制粒、整粒、干燥过程中羟丙纤维素和十二烷基硫酸钠混合溶液加入时,搅拌转速6rps,切刀转速6rps。
实施例4
参照实施例1-2的配方,依照表2的处方连续制备3批10万片的他达拉非片,其制备方法与实施例1相同,其处方如下表:
表2(10万片用量,各成分的计量单位为千克)
对比例1
处方与实施例1的相同,不同之处在于,他达拉非原料的微粉化过程中,不加入甘露醇和/或预胶化淀粉,而是直接将他达拉非原料药过60目筛,经气流粉碎处理1次,进料气压0.4mpa,粉碎气压0.2mpa,进行微粉,采用粒径检测仪对微粉化后的他达拉非进行检测。其他步骤都相同。
对比例2
制备工艺与实施例1相同,区别在于处方不同,主要是用淀粉替代预胶化淀粉,具体处方见下表3:
表3(1000单片用量及成分占比,各成分的计量单位为克)
对比例3
制备工艺与实施例相同,区别在于处方不同,主要是用乳糖替代甘露醇,具体处方见下表4:
表4(1000单片用量及成分占比,各成分的计量单位为克)
实验例1
对上述实施例1、2、3、4与对比例1~3的制备的他达拉非粉末组合物,采用马尔文粒径检测仪进行检测,其粒径d90如下表5。
表5实施例1~4与对比例1~3
从上述可以看出:他达拉非与甘露醇和/或预胶化淀粉一起进行微粉化,得到的微粒尺寸较小,达到d90≤10um,微粒在微粉化后,分散均匀。不加入甘露醇和/或预胶化淀粉(对比例1)、以其他稀释剂(淀粉)代替预胶化淀粉(对比例2)、以乳糖代替甘露醇(对比例3)、得到的他达拉非粉末组合物d90>10um微粒。
实验例2
对上述实施例1、2、3、4与对比例1~3的制备的他达拉非粉末组合物在水、空腹状态下的肠膜拟液(fassif)和餐后饱食状态下的肠膜拟液(fessif)介质中的进行渗透性实验,其结果如下表6:
表6实施例1~4与对比例1~3渗透性
实施例1~4多组试验数据表明,实施例制备的他达拉非粉末组合物在水、空腹状态下的肠膜拟液(fassif)和餐后饱食状态下的肠膜拟液(fessif)介质中渗透性大于或等于1.0x10-6cm/s,比对比例提高30%。对比例1他达拉非原粉未添加其他辅料,微粉化后的粉末,在水、空腹状态下的肠膜拟液(fassif)和餐后饱食状态下的肠膜拟液(fessif)介质中渗透性为0.5x10-6cm/s、0.6x10-6cm/s和0.6x10-6cm/s。对比例2和对比例3在水、空腹状态下的肠膜拟液(fassif)和餐后饱食状态下的肠膜拟液(fessif)介质中渗透性为0.6~0.7x10-6cm/s。
可见,本技术方案的他达拉分粉末组合物具有较高的渗透性,间接提高生物利用度。
实验例3
对实施例1-4制备得到的他达拉非片剂,进行溶出度检测。
他达拉非片剂溶出度按照如下方法检测:
照溶出度测定法(中国药典2010年版二部附录ⅹc第二法),以0.5%十二烷基硫酸钠溶液1000ml为溶出介质,转速为每分钟50转,依法操作;照高效液相色谱法(中国药典2010年版二部附录ⅴd),用辛烷基硅烷键合硅胶为填充剂;以甲醇-水(50:50)为流动相,检测波长为225nm,柱温40℃。精密量取对照品溶液50μl,注入液相色谱仪,记录色谱图,拖尾因子不大于1.5。精密量取供试品溶液与对照品溶液50μl,注入液相色谱仪,记录色谱图,按外标法以峰面积计算每片的溶出量。结果见表7。
表7实施例与对比例累积溶出
从上述可以看出,实施例1~4,累积溶出达到98%以上,对比例1~3累积溶出在96%以下,实施例1~4与对比例1~3体外溶出不一致。从体外的累积溶出可以间接的判断体内生物等效性,可见实施例1-4与对比例1-3体内的生物利用度将存在差异。
实验例4
对实施例1-4将制备得到的他达拉非片剂,进行杂质及含量的检测。
他达拉非片剂杂质和含量按照如下方法检测:
色谱条件:用辛烷基硅烷键合硅胶为填充剂;以乙腈-水-三氟乙酸(35:65:0.1)为流动相;检测波长为285nm;柱温为35℃。
杂质:取样品溶液10μl注入液相色谱仪,记录色谱图应不少于30分钟。样品溶液的色谱图中如有杂质峰,按面积归一化法计算,单个杂质峰面积不得大于总面积的0.2%,各杂质峰面积的和不得大于总面积的0.3%。
含量:精密称取样品(约相当于他达拉非25mg),置100ml量瓶中,加乙腈-水(50:50)使溶解并稀释至刻度,摇匀,滤过,精密量取续滤液10μl,注入液相色谱仪,记录色谱图;另精密称取他达拉非对照品约25mg,置100ml量瓶中,加乙腈-水(50:50)使溶解并稀释至刻度,摇匀,同法测定。按外标法以峰面积计算,即得。
结果见表8。
表8有关物质与含量检测数据
从表7、8中可以看出,实施例所制备的他达拉非片,具有总杂质<0.05%、含量高、溶出度好的优点。
实验例5
对实施例4制备的大批量(10万片)的他达拉非片剂与对照产品(希爱力)进行长期6个月的稳定性考察主要是考察总杂和含量的变化,结果见表9。
表9实施例4长期6个月的稳定性考察情况
实施例6
对实施例4制备的大批量(10万片)的他达拉非片剂进行比格犬生物利用度试验。采用市售的口服希爱力为对照产品。
他达拉非片剂生物利用度考察方法如下方法:
把12只比格犬随机分成4组,每组3只,进行双周期双交叉实验。实验前禁食12h,自由饮水。实验第一周期第1-3组比格犬分别口服实施例4-1~4-3制备的他达拉非片给药,第4组口服希爱力片给药,于给药后0.25,0.5,1,2,3,4,8,12,24,36和48h分别从前肢静脉取血约1000μl,置于肝素钠化离心管中,静置30min,5000rpm离心15min,分离血浆,置于-80℃冰箱冰冻保存,备用。经过一星期清洗期后,实验第二周期第1组比格犬口服希爱力片给药,第2-4组口服实施例4-1~4-3制备的他达拉非片给药,按上述步骤采血,分离血浆并冰冻保存。按照检测当天测定的随行标准曲线计算当天批次的所有血浆的血药浓度。结果见图1。
使用药动学计算软件,采用非房室模型-血管外给药模式对所测的药时曲线数据进行计算药动学参数,并进行统计学分析,见表10。
表10实施例4与原研药品(希爱力)药代动力学参数
由图1、表10可知,实施例4的他达拉非片剂的生物利用度对于市售的口服希爱力片一致并等效,其生物利用度在1.08-1.15之间。
- 还没有人留言评论。精彩留言会获得点赞!