一种提高熊果酸水溶性的共无定型复合物及其制备工艺的制作方法
本发明属于生物医药技术领域,具体涉及一种提高熊果酸水溶性的共无定型复合物及其制备工艺。
背景技术:
为解决药物的水难溶性和低溶解速率问题,许多制剂技术被应用,如微粉化、环糊精包合、固体分散体、加助溶剂和增溶剂、采用微乳剂和脂质体等新剂型、对药物进行成盐处理等方法。
通常药物的无定形态比晶态具有更高的溶解度,可以促进吸收,提高其口服生物利用度,但单个无定型药物稳定性差。固体分散制剂能使药物由晶体状态转变为无定形态,并维持较稳定的无定形态,溶解度和溶出速率增加,生物利用度得以提高。但聚合物固体分散体(polymericamorphoussoliddispersion,pasd)中药物在聚合物中的溶解性差,常会导致大量聚合物的加入,增加最终剂型的体积,此外大多数聚合物具有吸湿性,降低其稳定性。
在过去的几年里,共无定型药物系统(coamorphoussystems,cam)作为一种潜在的药物传递系统被提出并快速发展(rahulbchavanaetal,2016)。一种药物和一种小分子赋形剂(辅料)或者两种药物之间都可以形成二元的共无定型系统,cam作为一种有希望的替代pasd的方法,尤其以药物-药物cam的形式存在,可作为联合治疗药物使用,它不但可提高两种难溶性药物的水溶性及溶出速率,还能增强疗效和降低药物的毒副作用,部分体系甚至具有协同释药的潜力。
共无定型复合物的制备方法和工艺变量影响最终产品的结构特征,常用制备方法有熔融淬火法、溶剂挥发法、溶剂-喷雾(冷冻)干燥法和共研磨法等。制备共无定形药物时,要根据药物本身的性质,选择制备方法、组分比例及温度。
熊果酸为五环三萜类化合物,属于低溶解性和低渗透性药物,生物药剂学分类系统bcs(biopharmaceuticsclassificationsystem)中属于第四类药物。较高纯度的熊果酸为白色粉末,在乙醇中为白色针状结晶,存在溶解度差、口服吸收外排、生物利用度低、体内代谢速度快等生物药剂学问题。已有报道的提高熊果酸水溶性的制剂方法主要为聚合物固体分散体法,在提高水溶性和溶出度的同时,加入了大量聚合物,增加了最终剂型的体积,降低其稳定性。
技术实现要素:
本发明的目的在于提高熊果酸水溶性,克服聚合物固体分散体中因大量聚合物的加入,存在增加最终剂型的体积,以及聚合物具有吸湿性会降低其稳定性的缺陷,采用药物-药物共无定型系统,提供了一种提高熊果酸水溶性的共无定型复合物及其制备工艺。
为了实现上述目的,本发明采用的技术方案为:
一种提高熊果酸水溶性的共无定型复合物的制备工艺,其特征在于:其包括以下步骤:
1)将熊果酸加入无水乙醇中,水浴加热,搅拌,直至完全溶解,得到熊果酸醇溶液;
2)将槲皮素加入无水乙醇中,水浴加热,搅拌,直至完全溶解,得到槲皮素醇溶液;
3)按照熊果酸与槲皮素的质量比为1:0.02-0.2的比例,将槲皮素醇溶液倒入熊果酸醇溶中,混匀,旋转蒸发至乙醇全部挥发,然后将产物装入密封袋中保存,即得到所述共无定型复合物。
步骤1)所述水浴加热的温度为85-95℃,时间为15-20min;
步骤1)所述熊果酸醇溶液的浓度为1g/60ml。
步骤2)所述水浴加热的温度为45-55℃,时间为20-25min;
步骤2)所述懈皮素醇溶液的浓度为0.02-0.2g/140ml。
步骤3)所述熊果酸与槲皮素的质量比为1:0.05。
本发明所述制备的熊果酸-槲皮素共无定型复合物,性质稳定,为提高熊果酸等五环三萜类化合物水溶性提供了一个新的制备工艺。在制备方法上,本发明尝试了研磨法和溶剂法,由于研磨法效果较差,因此选用溶剂法制备;在溶剂选择上,本发明采用了甲醇与乙醇溶解,乙醇几乎没有毒性,对熊果酸溶解性较好,因此选用乙醇为溶剂;在制备比例上,选用了不同的熊果酸与槲皮素质量比,最终显示溶剂法以熊果酸与槲皮素质量比20:1时,熊果酸的水溶性提高最多,由原3.96mg/l提高至8.36mg/l,经粉末x射线衍射(pxrd)图谱分析和傅里叶变换红外光谱(ftir)分析,制备物表征显示为无定型复合物状态。
本发明的有益效果:本发明将两种具有活性的药物成分即熊果酸和槲皮素,通过制备药物-药物无定型系统的方法,以槲皮素作为配体(co-former),利用槲皮素玻璃态形成能力,与熊果酸形成稳定的无定型复合物,增加熊果酸的水溶性,以提高熊果酸的口服吸收及药效。方法简便易行,制备成的复合物没有增加过多的载体成分,解决了聚合物固体分散体中因大量聚合物的加入,增加最终剂型的体积,以及聚合物具有吸湿性会降低其稳定性的缺陷,同时提高了熊果酸的水溶性。
附图说明
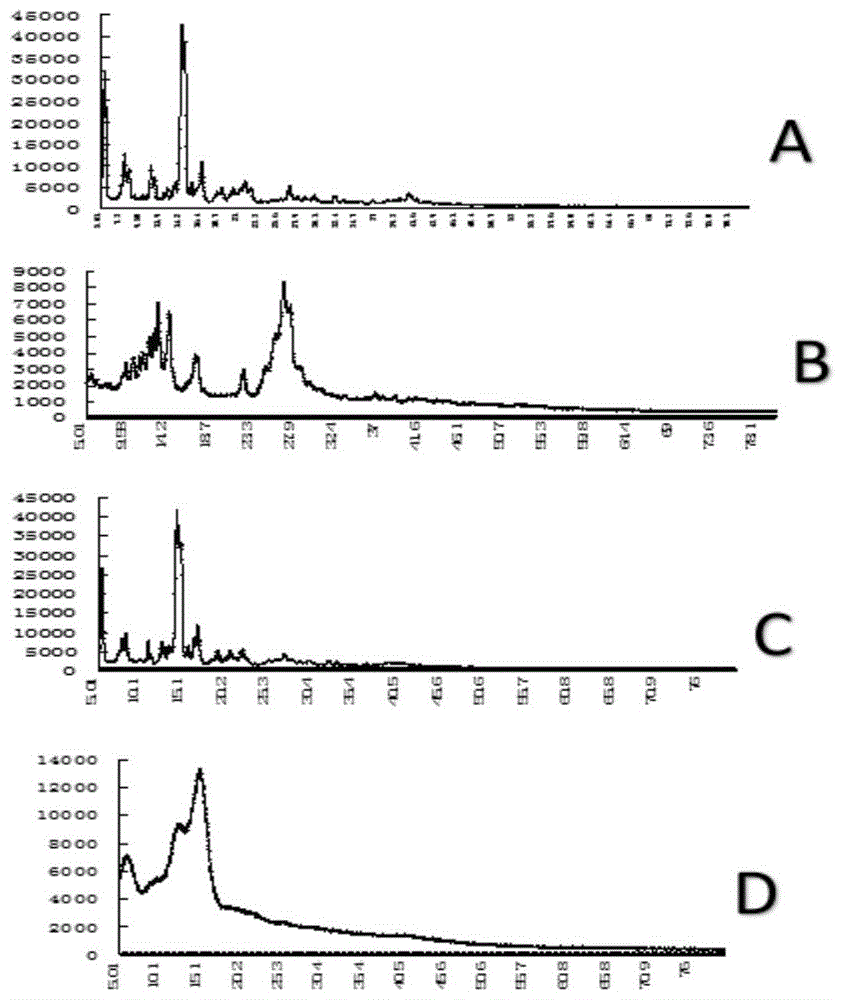
图1为实施例6的pxrd图,其中a为熊果酸单体、b为槲皮素单体、c为研磨法制备的熊果酸-槲皮素共无定型复合物,d为溶剂法制备的熊果酸-槲皮素复合物;
图2为实施例6的熊果酸ftir图;
图3为实施例6的槲皮素ftir图;
图4为实施例6的溶剂法制备的熊果酸-槲皮素共无定型复合物(1:0.05)ftir图;
图5为实施例6的研磨法制备的熊果酸-槲皮素复合物(1:0.2)ftir图。
具体实施方式
以下结合附图和具体实施方式对本发明作进一步详细的说明:
实施例1
熊果酸标准曲线的绘制
精密称取熊果酸标准品10mg,用无水乙醇定容于100ml容量瓶中,得到0.1mg/ml的熊果酸标准液。分别精密吸取熊果酸标准液0.5ml、0.6ml、0.7ml、0.8ml、0.9ml、1ml、1.1ml、1.2ml、1.3ml于具塞试管中,在水浴锅中进行水浴加热挥去溶剂后,加入5%的香草醛-冰醋酸0.5ml,高氯酸1.2ml,60℃水浴中加热15分钟,取出冰浴5分钟后移入试管中,加入乙酸乙酯4ml,摇匀后在波长550nm下测定其吸光度。以熊果酸含量作为横坐标,熊果酸吸光度作为纵坐标作标准曲线。标准曲线方程为y=9.51x-0.0348,r²=0.999。
实施例2
熊果酸水溶解度测定
取熊果酸标准品0.1g于500ml烧杯中,加入300ml蒸馏水,在60℃水浴中加热15分钟,期间不停地用玻璃棒搅拌。15分钟过后将烧杯放入90%功率超声波仪中进行超声10分钟,超声结束后再将烧杯放入60℃水浴锅中进行加热15分钟。最后将加热后的溶液进行旋转蒸发,直至溶液大约在50ml时,取出进行抽滤,量取滤液为所需的熊果酸水溶液。在波长550nm下测定其吸光度为0.342,经换算得水溶解度为3.96mg/l。
实施例3
溶剂法制备熊果酸-槲皮素共无定型复合物
取5个100ml的烧杯,每个烧杯分别放入精密称取的熊果酸标准品1g,加入60ml无水乙醇,在温度为90℃水浴锅中进行水浴加热15分钟,期间不停搅拌,直至熊果酸完全溶解,得到熊果酸醇溶液。
取5个250ml的烧杯,分别加入精密称取的槲皮素标准品0.02g、0.025g、0.05g、0.1g、0.2g,加入140ml的无水乙醇,在温度50℃的水浴锅中进行水浴加热20分钟,期间不停的搅拌,直至槲皮素完全溶解,得到懈皮素醇溶液。
往溶有0.02g、0.025g、0.05g、0.1g、0.2g的槲皮素醇溶液中分别倒入熊果酸醇溶液,先用玻璃棒不停地在烧杯中搅拌10分钟,后将混合液各自倒入蒸发瓶中进行旋转蒸发,直至乙醇全部挥发,将瓶内固体用钥匙刮取下,装入密封袋中,即得到熊果酸-槲皮素共无定型复合物。
实施例4
物理研磨法制备熊果酸-槲皮素复合物
分别取0.1g、0.2g、0.3g、0.4g、0.5g槲皮素分别置于5个研钵中,每研钵分别加入1g的熊果酸。往加入药品的研钵滴加10ml无水乙醇,并用研磨棒进行研磨1到2小时,期间乙醇挥发完后要继续添加。将研磨后的固体用密封袋装好,即得到熊果酸-槲皮素复合物。
实施例5
溶剂法与物理研磨法所得熊果酸-槲皮素复合物水溶解度测定
从实施例4各种比例的熊果酸-槲皮素复合物中取出0.1g分别放入500ml烧杯中,加入300ml水在60℃水浴锅中进行水浴加热15分钟,期间不停用玻璃棒搅拌;15分钟过后将烧杯放入90%功率超声波仪中进行超声10分钟,超声结束后再将烧杯放入60℃水浴锅中进行加热15分钟。将加热后的溶液倒入旋转蒸发瓶中进行旋转蒸发,旋转至50ml左右以保证溶液达到饱和状态,取出进行过滤,量取滤液体积为复合物水溶液体积。
从实施例3制备好的溶液中各取出10ml到50ml试管中在水浴锅中进行加热蒸干,加入5%香草醛-冰醋酸0.5ml,再加入1.2ml高氯酸,在60℃下水浴15分钟,将水浴后的溶液迅速放入冰水中冰浴5分钟使其终止反应,将冰浴后的溶液取出分别加入4ml乙酸乙酯,摇晃均匀后在波长为550nm下测定吸光度。
水溶解度测定结果如下:
熊果酸-槲皮素质量比为1:0.02、1:0.025、1:0.05、1:0.1、1:0.2溶剂法制备的熊果酸-槲皮素共无定型复合物,水溶解度分别为4.20、3.17、8.36、2..73、1.68mg/l,其中熊果酸-槲皮素质量比为1:0.05制备的复合物水溶解度最高,为单体熊果酸(水溶解度3.96mg/l)的2.11倍。
熊果酸-槲皮素质量比为1:0.1、1:0.2、1:0.3、1:0.4、1:0.5研磨法制备的熊果酸-槲皮素复合物,水溶解度分别为3.76、3.95、3.47、2.71、1.68mg/l,其中熊果酸-槲皮素质量比为1:0.2制备的复合物水溶解度最高。
溶剂法制备的熊果酸-槲皮素共无定型复合物效果比研磨法好。
实施例6
实施例3研磨法制备的熊果酸-槲皮素复合物(熊果酸-槲皮素质量比为1:0.05)与实施例4溶剂法制备的熊果酸-槲皮素共无定型复物(熊果酸-槲皮素质量比为1:0.2)的表征。
(1)粉末x射线衍射(pxrd)图谱分析
测试选取cu靶(40kv,40mv),2θ角,步进扫描0.026°/步,扫描范围5.00°~80.00°,每步停留时间36.465s,扫描速率4°/min。分别检测熊果酸单体、槲皮素单体、研磨法制备的熊果酸-槲皮素复合物,以及溶剂法制备的熊果酸-槲皮素共无定型复合物,相应的pxrd图见图1。
从pxrd图谱结果显示熊果酸衍射峰尖锐,在5.01°到16.4°有多个强的结晶衍射峰;槲皮素衍射峰尖锐,在5.01°到27.9°有多个强的结晶衍射峰;研磨法制备的熊果酸-槲皮素复合物的衍射峰尖锐,在5.01°到25.3之间有多个较强的结晶衍射峰,说明简单的物理混合未没有改变熊果酸的晶体状态;溶剂法制备的熊果酸-槲皮素共无定型复物的图谱中,复合物衍射峰较为粗钝,但是熊果酸与槲皮素二者的几个个明显衍射峰消失,说明熊果酸与槲皮素可能形成非晶型状态共存。
(2)傅里叶变换红外光谱(ftir)分析
采用kbr压片法测定ftir,将样品与kbr在干燥环境下混合制片后,分辨率为32cm-1,4000~400cm-1全谱扫描。分别检测熊果酸单体、槲皮素单体、研磨法制备的熊果酸-槲皮素复合物,以及溶剂法制备的熊果酸-槲皮素共无定型复物。
熊果酸单体扫描结果如图2所示,其中2个游离羟基3448,1个羰基1716,1701是olefinic的部位,9组ch2在2952(讯号强)。
槲皮素单体扫描结果如图3所示,其中5个游离羟基3385(讯号强),1个羰基1654,2组苯环(1617、1512)。
溶剂法制备的熊果酸-槲皮素共无定型复合物(1:0.05)扫描结果如图4所示,观测到共无定型复合物羟基讯号些微右移至3447,推测是游离羟基作为质子供体,参与氢键形成造成位移,并发现熊果酸的羰基些微左移至1717而槲皮素的羰基没有移动,仍在1654,但因比例较小,故讯号较弱。两组苯环讯号减弱,推测因槲皮素的羟基较熊果酸多且位于苯环周围,可能影响了共无定型复合物的苯环红外光吸收。羰基原分布于1654、1716,在共无定型复合物中位移至1717且峰形尖锐,也说明了羰基基团也参与了二聚体的结构形成。依据红外光谱的位移(羟基、羰基)显示,可以推测两分子间形成复合物,是通过氢键结合。
研磨法制备的熊果酸-槲皮素复合物(1:0.2)扫描图谱如图5所示。与溶剂法制备的共无定型复合物的红外光谱大致类似,唯有羟基部位不同,熊果酸的羟基讯号3448往左移至3524,而槲皮素的羟基讯号3385产生左移至3421,可见熊果酸与槲皮素在共无定型复合物与物理复合物之间,氢键的键结存在着明显的差异。结论:研磨法制备的复合物由其红外光谱发现,两个化合物羟基讯号分开,并未重合,是因为两分子之间没有产生氢键形成复合物,依旧是单一个体。
- 还没有人留言评论。精彩留言会获得点赞!