治疗食管癌的组合物及其应用的制作方法
本发明涉及四种治疗食管癌的活性单体及其组合物和应用,具体涉及从穿山龙、猪牙皂、土贝母和重楼四种中药中分别提取的活性单体,以及组成的治疗食管癌的组合物,属于中医药技术领域。
背景技术:
癌症医学术语称为恶性肿瘤,具有顽固性、转移性、危害性、死亡率高的特性,是21世纪主要的致死性非传染性疾病,给社会和家庭带来了沉重的经济负担。《2018年全球癌症统计报告》食管癌占所有恶性肿瘤的2%,居恶性肿瘤发病的第七位,总体死亡的第六位,其中男性患者占75%。食管癌的发生与亚硝胺慢性刺激、炎症与创伤、遗传因素以及饮水、食物和蔬菜中的微量元素含量有关,但确切原因还不明确。目前,食管癌的治疗方法主要有手术治疗、放射治疗、介入疗法、药物治疗等,但确诊时约有70-80%患者已属中晚期,大多数患者已失去最佳治疗时机,也使放射治疗、化学治疗收到很大的限制,不能有效地延长患者的生命及生活质量,且转移率高。
食管癌在中医学中属“噎膈”范畴,主要表现为饮食不下,膈咽不通,食则呕,其病位在食道,属胃气所主,与肝、脾、肾三脏功能密切相关,痰、热是最常见的食管癌发生发展的病因和病理因素。噎膈之症,必有顽痰,痰瘀生热,热毒日久耗伤阴血致血热,血热日久则化燥伤阴,治疗时以扶正补虚、清热活血、化痰散结为治则,以化痰、清热为大法。
穿山龙性平、味苦,归肝、肾、肺经,微寒清热、活血通络;猪牙皂,性温,味辛、咸,归肺、胃、大肠经,祛痰开窍、散结消肿;土贝母,性微寒,味苦,归肺、脾经,清热化痰,散结消肿;重楼,性微寒,味苦;归肝经;清热解毒,消肿止痛。研究表明,以上四种中药均具有抗肿瘤的作用。但由于现有中药组方种类较多,成分复杂,治病机理不明确,给中药推广应用造成了困难。目前没有针对抗食管癌的穿山龙、猪牙皂、土贝母和重楼中活性最强单体的报道,更没有出现活性单体组方的比例关系,药物对食管癌细胞作用机制的研究也不系统。
技术实现要素:
针对现有技术的不足,本发明的目的是提供一种从以穿山龙、猪牙皂、土贝母和重楼为原料,提取的活性单体及其组合物,以及其在治疗食管癌中的应用。
为了实现上述目的,本发明的技术方案之一是提供一种治疗食管癌的组合物,包括质量比为36:24:12:28~48:32:6:14的穿山龙活性单体、重楼活性单体、猪牙皂活性单体和土贝母活性单体。
优选的,该组合物包括质量比为42:28:9:21的穿山龙活性单体、重楼活性单体、猪牙皂活性单体和土贝母活性单体。
(a)穿山龙活性单体的制备方法:
(1)醇提:称取穿山龙,用95%(v/v)乙醇溶液常温浸泡后,抽滤,弃滤渣,浓缩,干燥得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中,然后正丁醇萃取,将萃取液旋蒸,干燥得正丁醇部位固体;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱,采用h2o-etoh体系洗脱,留取20%h2o-80%etoh部位洗脱液,干燥得20%h2o-80%etoh部位固体;
(4)ods层析柱分离:将20%h2o-80%etoh部位固体溶于超纯水中,通过ods层析柱,采用h2o-meoh体系洗脱,留取70%h2o-30%meoh部位洗脱液,干燥,得到淡黄色固体;
(5)硅胶层析柱分离:使用ch2cl2溶解淡黄色固体,湿法上样,采用etoac-meoh-h2o体系洗脱,留取洗脱液,干燥;
(6)纯化:使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到穿山龙活性单体。
(b)猪牙皂活性单体的制备方法:
(1)醇提:称取猪牙皂,用95%(v/v)乙醇溶液常温浸泡后,抽滤,弃滤渣,浓缩,干燥得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中,然后正丁醇萃取,将萃取液旋蒸,干燥得正丁醇部位固体;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱,采用h2o-etoh体系洗脱,留取50%h2o-50%etoh部位洗脱液,干燥得50%h2o-50%etoh部位固体;
(4)ods层析柱分离:将50%h2o-50%etoh部位固体溶于超纯水中,通过ods层析柱,采用h2o-meoh体系洗脱,留取30%h2o-70%meoh部位洗脱液,干燥,得到淡黄色固体;
(5)硅胶层析柱分离:使用ch2cl2溶解淡黄色固体,湿法上样,采用etoac-meoh-h2o体系洗脱,留取洗脱液,干燥;
(6)纯化:使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到猪牙皂活性单体;
(c)土贝母活性单体的制备方法:
(1)醇提:称取土贝母,用95%(v/v)乙醇溶液常温浸泡后,抽滤,弃滤渣,浓缩,干燥得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中,然后正丁醇萃取,将萃取液旋蒸,干燥得正丁醇部位固体;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱,采用h2o-etoh体系洗脱,留取30%h2o-70%etoh部位洗脱液,干燥得30%h2o-70%etoh部位固体;
(4)ods层析柱分离:将30%h2o-70%etoh部位固体溶于超纯水中,通过ods层析柱,采用h2o-meoh体系洗脱,留取20%h2o-80%meoh部位洗脱液,干燥,得到淡黄色固体;
(5)硅胶层析柱分离:使用ch2cl2溶解淡黄色固体,湿法上样,采用etoac-meoh-h2o体系洗脱,留取洗脱液,干燥;
(6)纯化:使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到土贝母活性单体。
(d)重楼活性单体的制备方法:
(1)醇提:称取重楼,用95%(v/v)乙醇溶液常温浸泡后,抽滤,弃滤渣,浓缩,干燥得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中,然后正丁醇萃取,将萃取液旋蒸,干燥得正丁醇部位固体;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱,采用h2o-etoh体系洗脱,留取10%h2o-90%etoh部位洗脱液,干燥得10%h2o-90%etoh部位固体;
(4)ods层析柱分离:将10%h2o-90%etoh部位固体溶于超纯水中,通过ods层析柱,采用h2o-meoh体系洗脱,留取40%h2o-60%meoh部位洗脱液,干燥,得到淡黄色固体;
(5)硅胶层析柱分离:使用ch2cl2溶解淡黄色固体,湿法上样,采用etoac-meoh-h2o体系洗脱,留取洗脱液,干燥;
(6)纯化:使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到重楼活性单体。
穿山龙和95%乙醇溶液的质量体积比为1g:7ml;猪牙皂和95%乙醇溶液的质量体积比为1g:7ml;土贝母和95%乙醇溶液的质量体积比为1g:7ml;重楼和95%乙醇溶液的质量体积比为1g:7ml。
步骤(a)-(2)、(b)-(2)、(c)-(2)和(d)-(2)中固体和超纯水的质量体积比为1g:50ml。
步骤(a)-(3)、和(d)-(3)中h2o-etoh体系为100%h2o、70%h2o-30%etoh、50%h2o-50%etoh、30%h2o-70%etoh、20%h2o-80%etoh、10%h2o-90%etoh、100%etoh;步骤(b)-(3)、(c)-(3)中h2o-etoh体系为100%h2o、70%h2o-30%etoh、50%h2o-50%etoh、30%h2o-70%etoh、100%etoh;步骤(a)-(3)、(b)-(3)、(c)-(3)和(d)-(3)中洗脱液体积为3倍柱体积。
步骤(a)-(4)、(b)-(4)、(c)-(4)和(d)-(4)中h2o-meoh体系为70%h2o-30%meoh、60%h2o-40%meoh、50%h2o-50%meoh、40%h2o-60%meoh、30%h2o-70%meoh、20%h2o-80%meoh、100%meoh,洗脱液体积为3倍柱体积。
步骤(a)-(5)和(b)-(5)中etoac-meoh-h2o体系中etoac、meoh、h2o的体积比为8:2:0.5;步骤(c)-(5)和(d)-(5)中etoac-meoh-h2o体系中etoac、meoh、h2o的体积比为7:5:0.5;步骤(a)-(5)、(b)-(5)、(c)-(5)和(d)-(5)中洗脱液体积为3倍柱体积。
步骤(a)-(6)中高效液相纯化的条件为vmeoh:vh2o=82:18,紫外吸收波长207nm,流速2ml/min;(b)-(6)中高效液相纯化的条件为vmeoh:vh2o=78:22,紫外吸收波长206nm,流速2ml/min;步骤(c)-(6)中高效液相纯化的条件为vmeoh:vh2o=75:25,紫外吸收波长265nm,流速2ml/min;步骤(d)-(6)中高效液相纯化的条件为vmeoh:vh2o=65:35,紫外吸收波长254nm,流速2ml/min。
本发明的技术方案之一是提供一种上述组合物在制备治疗食管癌药物中的应用。
本发明的有益效果:
本发明使用细胞模型,以活性追踪为导向,采用醇提、层析色谱及高效液相色谱等现代提取分离技术,从具有抗癌活性的穿山龙、猪牙皂、土贝母和重楼四种中药中分别提取活性单体,并使用化学谱图手段推测分子组成并确定各单体分子结构,以基线等比增减法确定单体用量比例,组合成治疗食管癌的组合物,然后将活性单体及组合物作用于多种食管癌细胞,研究其对食管癌的作用机制。本发明提供的四种活性单体及其组合物,均来源于天然药物的提取,属纯中药制剂,具有预防和治疗食管癌的作用,具有很大的市场潜力和广阔的市场应用前景。
附图说明
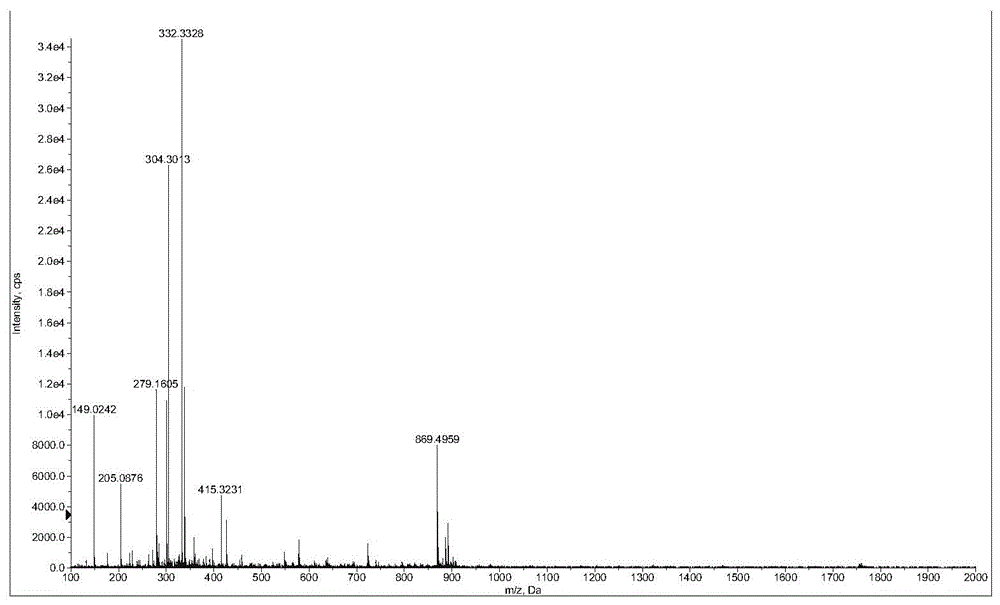
图1为穿山龙活性单体的质谱图。
图2为穿山龙活性单体的红外光谱图。
图3为猪牙皂活性单体的质谱图。
图4为猪牙皂活性单体的红外光谱图。
图5为土贝母活性单体的质谱图。
图6为土贝母活性单体的红外光谱图。
图7为重楼活性单体的质谱图。
图8为重楼活性单体的红外光谱图。
图9为穿山龙活性单体对四种食管癌细胞增殖的实时作用。
图10为猪牙皂活性单体对四种食管癌细胞增殖的实时作用。
图11为土贝母活性单体对四种食管癌细胞增殖的实时作用。
图12为重楼活性单体对四种食管癌细胞增殖的实时作用。
图13为组合物对四种食管癌细胞增殖的实时作用。
图14为活性单体和组合物对四种食管癌细胞的迁移的实时作用。图中,control为空白,csl为穿山龙,zyz为猪牙皂,tbm为土贝母,cl为重楼,com-4为组合物。
图15为活性单体和组合物作用于eca109细胞对细胞周期的影响。
图16为活性单体和组合物作用于细胞对ec9706细胞周期的影响。
图17为活性单体和组合物作用于te-1细胞对细胞周期的影响。
图18为活性单体和组合物作用于ec-1细胞对细胞周期的影响。
图19为活性单体和组合物对四种食管癌蛋白表达的影响。图中,con为空白,csl为穿山龙,zyz为猪牙皂,tbm为土贝母,cl为重楼,quan为组合物。
图20为hsa溶液随穿山龙活性单体溶液浓度增加对hsa的荧光猝灭的stern-volmer曲线(a:300k,b:310k)。
图21为hsa溶液随猪牙皂活性单体溶液浓度增加对hsa的荧光猝灭的stern-volmer曲线(a:300k,b:310k)。
图22为hsa溶液随土贝母活性单体溶液浓度增加对hsa的荧光猝灭的stern-volmer曲线(a:300k,b:310k)。
图23为hsa溶液随重楼活性单体溶液浓度增加对hsa的荧光猝灭的stern-volmer曲线(a:300k,b:310k)。
图24为hsa溶液随组合物溶液浓度增加对hsa的荧光猝灭的stern-volmer曲线(a:300k,b:310k)。
具体实施方式
以下结合实施例对本发明的具体实施方式作进一步详细说明。如无特殊说明,本发明所采用的实验方法为本领域的常规技术手段。
实施例1:穿山龙活性单体的制备
1.1提取方法
(1)醇提:称取穿山龙,用95%(v/v)乙醇溶液(质量体积比为1g:7ml)常温浸泡6天后,抽滤,弃滤渣,浓缩,真空-80℃冻干得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中(质量体积比为1g:50ml),在分液漏斗中依次使用石油醚、氯仿、乙酸乙酯、正丁醇等不同极性溶剂萃取,将五部位萃取液(石油醚、氯仿、乙酸乙酯、正丁醇、水)旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示穿山龙的正丁醇部位活性最强;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱hp20,采用水-乙醇(etoh)体系(100%h2o、70%h2o-30%etoh、50%h2o-50%etoh、30%h2o-70%etoh、20%h2o-80%etoh、10%h2o-90%etoh、100%etoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将七部位洗脱液旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示穿山龙20%h2o-80%etoh部位活性最强;
(4)ods层析柱分离:将20%h2o-80%etoh部位固体溶于超纯水中,通过ods层析柱(daisogelsp-120-50-ods-b),采用水-甲醇(meoh)体系(70%h2o-30%meoh、60%h2o-40%meoh、50%h2o-50%meoh、40%h2o-60%meoh、30%h2o-70%meoh、20%h2o-80%meoh、100%meoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将七部位洗脱液旋蒸,真空-80℃冻干得淡黄色固体,分别使用mtt法测试活性,结果显示70%h2o-30%meoh部位活性最强;
(5)硅胶层析柱分离:使用ch2cl2溶解70%h2o-30%meoh部位淡黄色固体,湿法上样,采用etoac(乙酸乙酯)-meoh-h2o体系(etoac、meoh、h2o的体积比为8:2:0.5)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,留取洗脱液,真空-80℃冻干得固体;
(6)纯化:依次使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到穿山龙活性单体。其中,葡聚糖凝胶分子筛:用甲醇溶解步骤(5)所得固体,加载到静置后的葡聚糖凝胶层析柱sephadexlh-20,采用70%h2o-30%meoh(v/v)流动相洗脱,洗脱液体积为3倍柱体积,流速为0.5ml/min,真空-80℃冻干得固体;重结晶:甲醇溶解所得固体,4.0μm滤头过滤后,逐滴滴加水至刚出现浑浊止,保鲜膜密封瓶口,使用5ml注射器针头在保鲜膜扎3×3共9个孔,静置观察固体出现,过滤留固体,再重复两次上述操作;高效液相纯化的条件为vmeoh:vh2o=82:18,紫外吸收波长207nm,流速2ml/min。
1.2穿山龙活性单体的化学结构鉴定
该物质为白色针状晶体;mp:276~278℃;可溶于水,易溶于甲醇、乙醇;元素分析,实测值:c,61.9%,h,8.2%,o,29.9%;计算值:c,62.2%,h,8.3%,o,29.5%,显示此化合物仅含c、h、o三种元素;质谱结果如图1,显示阳离子tof-msm/s:869[m+h]+,提示分子量为868;推测分子式c45h72o16。
红外光谱如图2,3600~3200cm-1区域出现一宽峰,归属为糖羟基,981,900,811cm-1处的三个螺旋结构特征峰,可判别25d系甾体皂苷,1639cm-1附近为c=c伸缩振动峰,1046cm-1附近为c-h平面变形振动峰。
核磁1h、13c谱表明,1h-nmr(500mhz,cd3od)在高场出现了δ0.77(3h,t,j=6.5hz,ch3-27),0.79(3h,s,ch3-18),0.96(3h,d,j=3.5hz,ch3-21),1.22(3h,s,ch3-19)四个甲基信号,烯氢信号δ5.37(1h,h-6);13c-nmr(125mhz,cd3od)谱13.5,15.4,16.1,16.5,16.6,18.4,20.6,28.5,29.4,30.0,31.0,31.3,31.4,31.8,36.6,37.2,38.1,39.5,40.0,41.5,47.1,47.3,47.4,47.5,47.8,47.9,48.1,50.3,56.4,60.5,62.3,66.5,68.4,69.3,70.8,71.0,71.1,72.3,72.5,75.2,76.6,77.9,78.6,80.8,99.0,100.9,101.6,109.2,121.2,140.5,给出三个糖端基碳信号δ101.6,100.9,99.3以及δ109.2的c-22季碳信号。因此,碳谱上45个碳信号中18个归属为糖部分,27个归属于皂苷元部分,烯碳信号δ140.4,121.3说明苷元c-5,6含有双键。
推测活性单体结构式为:
实施例2:猪牙皂活性单体的制备
2.1提取方法
(1)醇提:称取猪牙皂,用95%(v/v)的乙醇溶液(质量体积比为1g:7ml)常温浸泡6天后,抽滤,弃滤渣,浓缩,真空-80℃冻干得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中(质量体积比为1g:50ml),在分液漏斗中依次使用氯仿、乙酸乙酯、正丁醇等不同极性溶剂萃取,将四部位萃取液(氯仿、乙酸乙酯、正丁醇、水)旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示猪牙皂的正丁醇部位活性最强;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱hp20,采用水-乙醇(etoh)体系(100%h2o、70%h2o-30%etoh、50%h2o-50%etoh、30%h2o-70%etoh、100%etoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将五部位洗脱液旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示猪牙皂50%h2o-50%etoh部位活性最强;
(4)ods层析柱分离:将50%h2o-50%etoh部位固体溶于超纯水中,通过ods层析柱(daisogelsp-120-50-ods-b),采用水-甲醇(meoh)体系(70%h2o-30%meoh、60%h2o-40%meoh、50%h2o-50%meoh、40%h2o-60%meoh、30%h2o-70%meoh、20%h2o-80%meoh、100%meoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将七部位洗脱液旋蒸,真空-80℃冻干得淡黄色固体,分别使用mtt法测试活性,结果显示30%h2o-70%meoh部位活性最强;
(5)硅胶层析柱分离:使用ch2cl2溶解30%h2o-70%meoh部位淡黄色固体,湿法上样,采用etoac(乙酸乙酯)-meoh-h2o体系(etoac、meoh、h2o的体积比为8:2:0.5)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,留取洗脱液,真空-80℃冻干得固体;
(6)纯化:依次使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到猪牙皂活性单体。其中,葡聚糖凝胶分子筛:用甲醇溶解步骤(5)所得固体,加载到静置后的葡聚糖凝胶层析柱sephadexlh-20,采用30%h2o-70%meoh(v/v)流动相洗脱,洗脱液体积为3倍柱体积,流速为0.5ml/min,真空-80℃冻干得固体;重结晶:甲醇溶解所得固体,4.0μm滤头过滤后加入过量乙醚,静置析出固体,过滤留固体,再重复两次上述操作;高效液相纯化的条件为vmeoh:vh2o=78:22,紫外吸收波长206nm,流速2ml/min。
2.2猪牙皂活性单体的化学结构鉴定
该物质为白色无定型粉末;mp:309~311℃;易溶于水,甲醇,乙醇等溶剂;元素分析显示此化合物仅含c、h、o三种元素,实测值:c,59.6%,h,8.2%,o,32.2%;计算值:c,59.86%,h,8.24%,o,31.9%,质谱结果如图3,阳离子tof-msm/s:1004[m+h]+提示分子量为1003;推测分子式c50h82o20;红外光谱如图4,3600~3200cm-1区域出现一宽峰,归属为糖羟基。主要取代基的特征谱,1100~1010cm-1区域出现两个峰,表明分子中有呋喃糖苷,1719cm-1为羰基伸缩振动,2930cm-1为c-h非对称振动。
核磁1h、13c谱,1h-nmr(500mhz,cd3od)在高场出现了δ0.8~1.5之间有七个甲基信号。δ5.4是呋喃糖构型特征。12.5,16.2,17.1,17.9,18.3,19.5,24.4,24.6,26.4,27.9,28.5,31.6,33.3,33.5,37.9,40.3,40.7,40.8,41.8,43.0,57.0,64.3,66.8,67.2,67.3,68.6,69.7,70.9,71.1,71.8,71.9,73.2,73.6,75.1,75.7,75.9,76.8,77.5,77.7,78.1,81.5,87.9,103.5,105.8,106.6,106.7,112.5,112.6,13c-nmr(125mhz,cd3od)谱给出四个糖端基碳信号δ103.0,105,106,106,分别为呋喃糖、葡萄糖和木糖,δ78.1说明骨架为齐墩果酸。
推测活性单体结构式为:
实施例3:土贝母活性单体的制备
3.1提取方法
(1)醇提:称取土贝母,用95%(v/v)的乙醇溶液(质量体积比为1g:7ml)常温浸泡6天,抽滤,弃滤渣,浓缩,真空-80℃冻干得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中(质量体积比为1g:50ml),在分液漏斗中依次使用氯仿、乙酸乙酯、正丁醇等不同极性溶剂萃取,将四部位萃取液(氯仿、乙酸乙酯、正丁醇、水)旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示土贝母的正丁醇部位活性最强;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱hp20,采用水-乙醇体系(100%h2o、70%h2o-30%etoh、50%h2o-50%etoh、30%h2o-70%etoh、100%etoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将五部位洗脱液旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示土贝母30%h2o-70%etoh部位活性最强;
(4)ods层析柱分离:将30%h2o-70%etoh部位固体溶于超纯水中,通过ods层析柱(daisogelsp-120-50-ods-b),采用水-甲醇体系(70%h2o-30%meoh、60%h2o-40%meoh、50%h2o-50%meoh、40%h2o-60%meoh、30%h2o-70%meoh、20%h2o-80%meoh、100%meoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将七部位洗脱液旋蒸,真空-80℃冻干得淡黄色固体,分别使用mtt法测试活性,结果显示土贝母20%h2o-80%meoh部位活性最强;
(5)硅胶层析柱分离:使用ch2cl2溶解20%h2o-80%meoh部位淡黄色固体,湿法上样,采用etoac-meoh-h2o体系(etoac、meoh、h2o的体积比为7:5:0.5)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,留取洗脱液,真空-80℃冻干得固体;
(6)纯化:依次使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到土贝母活性单体;其中,葡聚糖凝胶分子筛:用甲醇溶解步骤(5)所得固体,加载到静置后的葡聚糖凝胶层析柱sephadexlh-20,采用20%h2o-80%meoh(v/v)流动相洗脱,洗脱液体积为3倍柱体积,流速为0.5ml/min,真空-80℃冻干得固体;重结晶:甲醇溶解所得固体,4.0μm滤头过滤后,逐滴滴加水至刚出现浑浊止,保鲜膜密封瓶口,使用5ml注射器针头在保鲜膜扎3×3共9个孔,静置观察固体出现,过滤留固体,再重复两次上述操作;高效液相纯化的条件为vmeoh:vh2o=75:25,紫外吸收波长265nm,流速2ml/min。
3.2土贝母活性单体的化学结构鉴定
该物质为无定型白色粉末;mp:261~263℃;微溶于水,易溶于甲醇、乙醇。元素分析,实测值:c,56.5%,h,7.3%,o,36.2%;计算值:c,56.3%,h,7.3%,o,36.4%,表明此化合物仅含c、h、o三种元素。质谱结果如图5,阳离子tof-msm/s:1388.5[m+na]+提示分子量为1365.3;推测分子式c64h100o31。
红外光谱如图6,3600~3200cm-1区域出现一宽峰,归属为糖羟基。主要取代基的特征谱,1039、1131cm-1区域出现两个峰,表明分子中有c-o键的伸缩振动,3385cm-1为羟基的伸缩振动,1653cm-1为羰基伸缩振动,2934cm-1为c-h非对称伸缩振动。
核磁1h、13c谱结果,1h-nmr(500mhz,cd3od)在高场出现了δ0.77(3h,t,j=6.5hz,ch3-27),0.79(3h,s,ch3-18),0.96(3h,d,j=3.5hz,ch3-21),1.22(3h,s,ch3-19)四个甲基信号,烯氢信号δ5.37(1h,h-6)。碳谱15.2,17.6,17.9,18.0,19.2,24.6,26.1,27.3,31.3,32.6,33.4,34.2,36.6,37.1,37.6,40.9,41.6,42.7,42.8,46.1,46.4,47.7,48.2,48.4,49.9,62.2,64.4,64.8,67.0,68.0,68.6,70.4,70.6,70.7,70.9,70.9,72.9,73.6,74.0,74.8,75.8,76.4,76.7,77.3,77.4,77.5,78.3,84.0,84.7,95.1,102.1,103.5,105.7,106.2,123.5,145.1,172.3,172.5,176.8,
13c-nmr(125mhz,cd3od)谱给出三个糖端基碳信号δ103.0,102.3,100.4以及δ110.5的c-22季碳信号。因此,碳谱上45个碳信号中18个归属为糖部分,27个归属于皂苷元部分,烯碳信号δ145.1,123.5说明苷元c-5,6含有双键。
推测活性单体结构式为:
实施例4:重楼活性单体的制备
4.1提取方法
(1)醇提:称取重楼,用95%(v/v)乙醇溶液(质量体积比为1g:7ml)常温浸泡6天,抽滤,弃滤渣,浓缩,真空-80℃冻干得棕黄色固体;
(2)萃取:将棕黄色固体溶于超纯水中(质量体积比为1g:50ml),在分液漏斗中依次使用石油醚、氯仿、乙酸乙酯、正丁醇等不同极性溶剂萃取,将五部位萃取液(石油醚、氯仿、乙酸乙酯、正丁醇、水)旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示重楼的正丁醇部位活性最强;
(3)大孔树脂层析分离:正丁醇部位固体溶于超纯水中,通过大孔树脂层析柱hp20,采用水-乙醇体系(100%h2o、70%h2o-30%etoh、50%h2o-50%etoh、30%h2o-70%etoh、20%h2o-80%etoh、10%h2o-90%etoh、100%etoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将七部位洗脱液旋蒸,真空-80℃冻干得固体,分别使用mtt法测试活性,结果显示重楼10%h2o-90%etoh部位活性最强;
(4)ods层析柱分离:将10%h2o-90%etoh部位固体溶于超纯水中,通过ods层析柱(daisogelsp-120-50-ods-b),采用水-甲醇体系(70%h2o-30%meoh、60%h2o-40%meoh、50%h2o-50%meoh、40%h2o-60%meoh、30%h2o-70%meoh、20%h2o-80%meoh、100%meoh,v/v)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,将七部位洗脱液旋蒸,真空-80℃冻干得淡黄色固体,分别使用mtt法测试活性,结果显示重楼40%h2o-60%meoh部位活性最强;
(5)硅胶层析柱分离:使用ch2cl2溶解40%h2o-60%meoh部位淡黄色固体,湿法上样,采用etoac-meoh-h2o体系(etoac、meoh、h2o的体积比为7:5:0.5)洗脱,洗脱液体积为3倍柱体积,流速为1ml/min,留取洗脱液,真空-80℃冻干得固体;
(6)纯化:依次使用葡聚糖凝胶分子筛、重结晶及高效液相纯化,得到重楼活性单体。其中,葡聚糖凝胶分子筛:用甲醇溶解步骤(5)所得固体,加载到静置后的葡聚糖凝胶层析柱sephadexlh-20,采用10%h2o-90%meoh(v/v)流动相洗脱,洗脱液体积为3倍柱体积,流速为0.5ml/min,真空-80℃冻干得固体;重结晶:甲醇溶解所得固体,4.0μm滤头过滤后,逐滴滴加水至刚出现浑浊止,保鲜膜密封瓶口,使用5ml注射器针头在保鲜膜扎3×3共9个孔,静置观察固体出现,过滤留固体,再重复两次上述操作;高效液相纯化的条件为vmeoh:vh2o=65:35,紫外吸收波长254nm,流速2ml/min。
4.2重楼活性单体的化学结构鉴定
该物质为白色无定型粉末;mp:212~214℃;微溶于水,易溶于甲醇、乙醇。元素分析,实测值:c,61.9%,h,8.3%,o,29.8%,计算值:c,61.80%,h,8.25%,o,29.95%,表明此化合物仅含c、h、o三种元素,质谱结果如图7,阳离子tof-msm/s:877.3[m+na]+提示分子量为854.3;推测分子式c44h70o16。
红外光谱如图8,在3700~3200cm-1区域出现一宽峰,属于o-h的伸缩振动,归属为糖羟基。主要取代基的特征谱,3000~2800cm-1区域出现两个峰,归属为饱和c-h的伸缩振动,1452cm-1为-ch3的不对称变形振动,1378cm-1为-ch3的对称变形振动。
核磁1h、13c谱,1h-nmr(500mhz,cd3od)在高场出现了δ0.77(3h,t,j=6.5hz,ch3-27),0.79(3h,s,ch3-18),0.96(3h,d,j=3.5hz,ch3-21),1.22(3h,s,ch3-19)四个甲基信号,烯氢信号δ5.37(1h,h-6)。碳谱14.9,16.8,17.5,17.9,18.0,18.6,19.8,22.0,29.9,30.7,31.4,32.4,32.7,33.2,38.0,38.6,40.9,41.4,42.9,51.7,57.8,63.0,63.7,67.9,69.7,72.4,73.9,76.5,77.8,78.0,79.3,82.2,83.2,85.9,100.5,102.1,109.9,110.6,122.6,141.9,13c-nmr(125mhz,cd3od)谱给出三个糖端基碳信号δ109.9,102.1,100.5以及δ110.6的c-22季碳信号。因此,碳谱上44个碳信号中17个归属为糖部分,27个归属于皂苷元部分,烯碳信号δ141.9,122.6说明苷元c-5,6含有双键。
推测活性单体结构式为:
实施例5:治疗食管癌的组合物
利用mtt法检测穿山龙活性单体、猪牙皂活性单体、土贝母活性单体和重楼活性单体对常见食管癌细胞(eca109、ec9706、te-1、ec-1)的作用,具体方法为:
1)将对数生长期细胞进行消化、离心、重悬、计数,接种于96孔板中,3个复孔,1.0×104cells/孔,200μl/孔,eca109、te-1、ec-1细胞使用rpmi1640培养基培养,ec9706使用dmem培养基培养,培养基中含10%fbs,本发明细胞所用培养基遵循此规则;
2)培养箱孵育,5%co2、37℃、饱和湿度,24h后取出,弃上层培养液,设置空白组和加药组,空白组更换含10%fbs培养基,加药组更换含工作浓度药物的10%fbs培养基,工作浓度设置8个浓度梯度,100μg/ml、50μg/ml、25μg/ml、12.5μg/ml、6.25μg/ml、3.125μg/ml、1.563μg/ml,0.781μg/ml,200μl/孔;
3)培养箱孵育,5%co2、37℃、饱和湿度,48h后取出,弃上清,加mtt工作液100μl/孔,培养箱内孵育4h,弃上清,加入dmso150μl/孔,轻叩96孔板周边,摇床摇15min;
4)酶标仪570nm、630nm双波长测定od(吸光度)值;
5)计算抑制率。抑制率(%)=(1-加药组od值/空白组od值)×100%;
6)使用spssstatistics17.0软件分析系统对实验结果进行数据分析,得ic50值。
结果显示,四种活性单体对四种食管癌细胞均有明显的生长抑制作用,穿山龙活性单体对eca109、ec9706、te-1、ec-1四种食管癌细胞作用的ic50值分别为1.29±0.12μg/ml、1.41±0.02μg/ml、3.72±0.28μg/ml、1.11±0.14μg/ml;猪牙皂活性单体对eca109、ec9706、te-1、ec-1四种食管癌细胞作用的ic50值分别为2.42±0.09μg/ml、5.94±0.45μg/ml、35.58±0.58μg/ml、3.56±0.28μg/ml;土贝母活性单体对eca109、ec9706、te-1、ec-1四种食管癌细胞作用的ic50值分别为1.93±0.09μg/ml、1.97±0.09μg/ml、7.90±0.41μg/ml、1.23±0.02μg/ml;重楼活性单体对eca109、ec9706、te-1、ec-1四种食管癌细胞作用的ic50值分别为3.04±0.28μg/ml、0.63±0.04μg/ml、1.85±0.09μg/ml、0.61±0.02μg/ml。
基线等比增减设计法是针对a、b两种药物总量恒定的前提下,以一定的配比为基线,a药含量以10%~30%递减,b药含量以10%~30%递增,或反之,向两侧扩增,直至扩大到极点,两侧极点分别为单纯a药和单纯b药,基线等比增减试验设计法试验适合二元药物的组合配比设计。
由于基线等比增减设计法适合确定组合物中二元的比例关系,故将四活性单体事先分为二组,使用此法确定各组中两种活性单体的比例关系,再将二组作为二元,使用本法确定比例关系,进而得到四活性单体比例。本发明所涉及四味中药,其药性分别为穿山龙归肝、肺经,可清热祛风、活血通络,猪牙皂归肺、大肠经,可祛痰开窍,散结消肿,土贝母归肺、脾经,可散结消痈、利痰,重楼归肝经,可清热解毒,消肿止痛。四味药物中,穿山龙和重楼均具有清热功效,且活性单体均具有甾体皂苷骨架,故将其归为一组。猪牙皂和土贝母均具有化痰之功效,且活性单体均具有多氢蒎五环母核,故将其归为一组。
本发明以质量比5:5为基线,两侧以10%递变,a:b分别为10:0、9:1、8:2、7:3、6:4、5:5、4:6、3:7、2:8、1:9、0:10等,分别作用于四种食管癌细胞,同时设置对照组,通过mtt法检测抑制率,得到最佳配比。结果表明,在猪牙皂活性单体和土贝母活性单体的一组中,当mzj:mtbm=3:7时组合物对四种食管癌细胞的增殖抑制能力最强,故选取3:7作为组合物中猪牙皂活性单体和土贝母活性单体的最佳质量比;在穿山龙活性单体和重楼活性单体的一组中,当mcsl:mcl=6:4时组合物对四种食管癌细胞的增殖抑制能力最强,故选取6:4作为组合物中穿山龙活性单体和重楼活性单体的最佳组合比例;在两组组合时,当mqing:mhua=6:4-8:2时组合物对四种食管癌细胞具有较强的增殖抑制能力,其中当mqing:mhua=7:3时组合物对四种食管癌细胞的增殖抑制能力最强,故选取7:3作为组合物中甾体皂苷类活性单体和多氢蒎五环母核类活性单体的最佳组合比例,进而得到四活性单体比例为,mcsl:mcl:mzj:mtbm=42:28:9:21,后续试验采用该配比的组合物(表1)。
表1.甾体皂苷与多氢蒎五环母核类活性单体二元作用对四种食管癌细胞的抑制率(n=3,
注:zj和tbm分别表示猪牙皂活性单体和土贝母活性单体,csl和cl分别表示穿山龙活性单体和重楼活性单体,qing和hua分别表示甾体皂苷类和多氢蒎五环母核类,p<0.05。
实施例6:活性单体及组合物对四种食管癌细胞的作用
6.1rtca实验测试细胞增殖
试验具体操作方法参照xcelligencertcadp系统基本实验操作protocol。将eca109、ec9706、ec-1、te-1四种食管癌细胞分别种植于e-plate16孔板,种板24h后加药,加药作用48h停止实验,实验设置空白组(不加药)和加药组,加药组设置7个浓度梯度,将3000μg/ml药物母液(活性单体和组合物)倍比稀释共7个浓度,分别为3000μg/ml、1500μg/ml、750μg/ml、375μg/ml、187.5μg/ml、93.75μg/ml、46.875μg/ml,加药时取5μl加入对应孔,此时终浓度分别为100μg/ml、50μg/ml、25μg/ml、12.5μg/ml、6.25μg/ml、3.125μg/ml、1.5625μg/ml,以此浓度的含药血清培养基作用于细胞,测定细胞指数cellindex,即ci,通过细胞指数的大小体现药物对细胞的作用影响。以时间为横轴,ci为纵轴,各药物作用不同细胞结果如图9-13。
图9为穿山龙活性单体对四种食管癌细胞的实时作用,与空白组(control)相比,7种浓度梯度的穿山龙活性单体均能降低细胞的ci值,且呈浓度梯度的依赖关系,即随着浓度的增加其ci值降低,对于不同的细胞其药物作用的敏感程度也有差异。
图10为猪牙皂活性单体对四种食管癌细胞的实时作用,与空白组(control)相比,7种浓度梯度的猪牙皂活性单体均能降低四种细胞的ci值,且呈浓度梯度的依赖关系。猪牙皂活性单体对于不同的细胞其药物作用的敏感程度存在差异,其中eca109最敏感,te-1较其它三种细胞不敏感。
图11为土贝母活性单体对四种食管癌细胞的实时作用,与空白组(control)相比,7种浓度梯度的土贝母活性单体均能降低四种细胞的ci值,且呈浓度梯度的依赖关系。四种食管癌细胞中,te-1弱于其它三种细胞对土贝母活性单体的敏感性。
图12为重楼活性单体对四种食管癌细胞的实时作用,与空白组(control)相比,各种浓度梯度的重楼活性单体均能降低四种细胞的ci值,且随浓度的增大、时间的增长抑制能力增强,存在时间和浓度的依赖关系,加药后6h(24-30h)内,药物作用基本稳定。
图13为组合物对四种食管癌细胞的实时作用,与空白组(control)相比,7种浓度梯度的组合物均能降低细胞的ci值,且呈浓度梯度的依赖关系,即随着浓度的增加其ci值降低。较空白组,加药组均能抑制食管癌细胞的增殖,且存在时间和浓度的依赖关系,加药后6h(24-30h)内,药物作用基本稳定。
6.2rtca实验测试细胞迁移
1)细胞悬液、药物配制步骤与6.1rtca实验同;
2)迁移板分上室和下室。超净台中取出16孔板,上室于plate上,蓝色标志点位于夹具的左上方,下室加工作液。空白组165μl/孔,含10%fbs培养基,加药组165μl/孔,含10%fbs、ic50值药物浓度的培养基,3个复孔。操作过程避免产生气泡,操作后下室液面呈向上凸出弧形。将上下室标志点对准按压扣上,30μl/孔向上室里加不含血清培养基,培养箱中平衡1h。
3)将平衡后的迁移板置于培养箱中的rtca-station上,检测连接状态,设置相关参数,具体步骤与rtca细胞增殖类似。
4)平衡1h后,上室加入细胞悬液,100μl/孔,加药组额外加入ic50浓度的药物悬液,5μl/孔。迁移板放置超净台内静置30min,待细胞沉降后,放置到培养箱中的rtca-station上,设定检测时间48h,采集、储存实验数据。
结果见图14。由图可知,与空白组相比,加药组ci值明显降低,表明四活性单体及组合物均能降低四细胞的迁移能力,且对于同种细胞,活性单体和组合物对其迁移能力的降低水平存在差异,组合物较单一单体对四种细胞的影响更为显著。
6.3细胞克隆
6.3.1主要试剂的配制
琼脂糖底层琼脂和上层琼脂,其质量浓度分别为1.2%,0.7%,配制方法类似,称取1.2g,0.7g琼脂糖粉分别溶于100ml生理盐水中,高压灭菌,4℃冰箱储存备用。
6.3.2软琼脂克隆操作步骤
1)将1.2%的软琼脂和2×完全培养基(含20%fbs)各30ml等体积混合,制成0.6%底层琼脂,铺于24孔板中,0.8ml/孔,19.2ml/板,室温静置凝固。
2)将对数生长期食管癌细胞eca109、ec9706、ec-1、te-1进行消化、重悬、计数,分别制成细胞悬液。
3)将0.7%的软琼脂和2×完全培养基(含20%fbs)各30ml等体积混合,制成0.35%上层琼脂,分别加入细胞悬液至细胞浓度为250cells/ml,设置空白组(不加药)和加药组,加药组加入药物至药物浓度为ic50值,混匀,0.8ml/孔,19.2ml/板,每孔细胞数200个,超净台室温静置凝固,置培养箱中孵育,22天后观察、计数。
4)设置3个复孔,实验重复3次,取均值。
6.3.3细胞集落形成率
细胞集落形成率=平均细胞集落数/每孔接种细胞数×100%
6.3.4统计学分析
使用spssstatistics17.0软件分析系统对实验结果进行数据分析,以均数±标准差
6.3.5实验结果
空白组和加药组对4种食管癌细胞处理后,22天后观察与空白组相比,加药组克隆细胞群落变小,形态不规则,结果如表2所示:
表2.活性单体及组合物对4种细胞作用后克隆形成的集落数(n=3,x±s)
表2可知,对于4种食管癌细胞,与空白组比较,四活性单体及组合物均能抑制细胞集落的形成,细胞集落形成数量出现明显下降,不同的单体对于不同的细胞存在明显差异,对于eca109细胞加药组的集落形成较空白组减少1/2~2/3之间,其他三种细胞加药组较空白组细胞集落数目减少1/3~1/2之间。就细胞集落形成率而言,空白组ec9706、eca109、te-1、ec-1细胞大约在21.7%~34.3%之间,加药组大约在7.9%~24.4%之间,四种细胞的加药组集落形成率较空白组均有所下降,其下降范围在9.7~26.1个百分点之间。
6.4细胞周期
将对数生长期食管癌细胞eca109、ec9706、ec-1、te-1进行消化,离心,重悬,计数,接种于100mm培养皿中,106cells/皿,10ml/皿,培养箱内培养24h,吸取上清,空白组加含10%fbs培养基,加药组加穿山龙活性单体、猪牙皂活性单体、土贝母活性单体、重楼活性单体和组合物ic50值药物浓度的含10%fbs培养基,10ml/皿,每组3皿,继续培养48h。观察细胞形态,拍照。各皿弃上清,预冷pbs洗涤2次,0.125%不含edta消化胰酶消化,倒置显微镜观察细胞收缩,变圆,部分脱壁,0.5mlfbs终止消化,1500rpm离心7min,弃上清,预冷pbs洗涤2次,计数,取106cells于流式管中,1500rpm离心7min,弃上清,加入预冷无水乙醇,调整乙醇终浓度为70%,乙醇固定24h,1500rpm离心7min,弃上清,预冷pbs洗涤2次,1ml/次,使用蛋白周期测定试剂盒,分别加入pi(碘化丙啶)/rnaase500μl,4℃避光30min,300目尼龙过滤,流式细胞仪上样测试,开启cellquestpro软件,设置激发波长488nm,采集细胞荧光信号20000events,采集结束modfit软件分析各期分布,结果见图15-18。
图15为活性单体和组合物作用于eca109细胞对细胞周期的影响。与空白组相比,四活性单体及组合物对食管癌eca109作用后,g0/g1期细胞比率由35.07%变为45.08%~61.62%,均显著增加,s期细胞比率由58.15%降低到35.72%~49.06%之间,说明药物把细胞分裂阻滞在dna合成前期,且组合物对eca109细胞周期的影响较活性单体显著。
图16为活性单体和组合物作用于ec9706细胞对细胞周期的影响。与空白组相比,四活性单体及组合物对食管癌ec9706细胞周期均有改变,药物作用ec9706后,猪牙皂活性单体、土贝母活性单体、重楼活性单体和组合物作用后细胞g0/g1期细胞比率增加,s期及g2/m期细胞比率降低,穿山龙活性单体对细胞的分裂阻滞有所差异。
图17为活性单体和组合物作用于te-1细胞对细胞周期的影响。与空白组相比,四活性单体和组合物对食管癌te-1细胞周期影响存在较大差异,穿山龙活性单体、猪牙皂活性单体和组合物作用te-1后,g0/g1期细胞比率减少。重楼活性单体和土贝母活性单体作用te-1细胞后,g0/g1期细胞比率增加,而细胞s期细胞比率降低。
图18为活性单体和组合物作用于ec-1细胞对细胞周期的影响。与空白组相比,四活性单体和组合物均对食管癌ec-1细胞周期产生显著影响,g0/g1期细胞比率均增加,g2/m期细胞比率降低。
6.5westernbloting
6.5.1抗体及操作步骤
所用一抗为rbmabtoegfr,momabplcγ1,rbmabtopkcalpha,rbpabtobetaactin,使用时1:2000稀释。二抗为hrbcoatanti-rabbitigg,hrbgoatanti-mouseigg使用时1:1500稀释。操作步骤参照《分子生物学实用实验技术》第二章western-blot技术。
6.5.2实验结果
以ic50药物浓度,将四活性单体和组合物分别作用于四种食管癌细胞,提取蛋白、定量、8%sdspage胶分离、湿转、一抗孵育、二抗孵育然后化学发光、暗室显影得四种细胞的egfr、plc-γ1和pkcα蛋白表达(见图19)。如图19所示,对蛋白表达各条带进行分析,经药物作用后,较空白组,四种食管癌细胞的egfr、plc-γ1和pkcα三种蛋白表达均降低,重楼活性单体较穿山龙活性单体对plc-γ1和pkcα蛋白影响强,土贝母活性单体较猪牙皂活性单体对三蛋白影响强,组合物对各蛋白的影响强度强于各活性单体。以上结果表明,不同活性单体不同程度抑制四种食管癌细胞的三种目标蛋白的表达,但各活性单体对不同细胞的中的不同蛋白表达存在较大的差异性。
实施例7:活性单体和组合物与人血清白蛋白(hsa)的相互作用
7.1溶液的配制
tris-hcl缓冲溶液的配制:分别称取tris-base3.0292g、nacl7.3102g于250ml容量瓶中,双蒸水溶解后定容,浓hcl调ph至7.32,4℃保存备用。
hsa溶液配制:以双蒸水为溶剂,配成浓度为5×10-5mol/l的hsa溶液,过夜,4℃保存备用。
四活性单体及组合物样品溶液的配制:分别称取穿山龙活性单体5.06mg、猪牙皂活性单体6.01mg、土贝母活性单体8.18mg、重楼活性单体5.13mg、组合物6.15mg,以甲醇为溶剂,制备10-3mol/l样品溶液,4℃保存备用。
7.2荧光光谱测定
实验组为10mlhsa(1×10-6mol/l)溶液,由9.80mltris-hcl缓冲溶液和0.20mlhsa(5×10-5mol/l)溶液配制,空白组为纯tris-hcl溶液。实验时依次加入不同体积的活性单体和组合物样品溶液,改变样品溶液浓度,研究不同活性单体和组合物浓度对has内源性荧光强度的影响。实验参数设置:λex=280nm,exslit=5nm,emslit=5nm,扫描范围285nm-450nm。设δλ=60nm或δλ=15nm,扫描同步荧光光谱。实验温度300k、310k。
所用荧光分光光度计为日本hitachi公司的f-7000荧光分光光度计。
7.3实验结果
7.3.1与hsa的相互作用的发射光谱
在300k和310k,ph=7.32,tris-hcl缓冲溶液,hsa(1×10-6mol/l)溶液,激发波长(λex)为280nm条件下,随活性单体和组合物样品溶液浓度的增加,hsa中的内源性荧光强度均逐渐减弱,表明样品溶液可以猝灭hsa的内源性荧光,但其最大发射峰的形状及位置基本不变,样品溶液与hsa之间发生了较强的相互作用,推测形成荧光较弱的复合物造成hsa内源性荧光猝灭。组合物(300k)随溶液浓度的增加,hsa的内源性荧光强度逐步上升,表明此温度下组合物溶液可以增强hsa的内源性荧光,但其最大发射峰的形状及位置基本不变。
7.3.2与has相互作用的同步荧光光谱
温度为300k和310k,ph=7.32,tris-hcl缓冲溶液,δλ=15nm,hsa(1×10-6mol/l),穿山龙活性单体、猪牙皂活性单体、土贝母活性单体、重楼活性单体和组合物随着药物浓度的增加,hsa酪氨酸残基最大吸收峰的强度逐渐升高,但其峰位基本不变,表明样品溶液对蛋白质分子构象的改变不大。
在温度为300k和310k时,ph=7.32,tris-hcl缓冲溶液,δλ=60nm,随药物浓度增加,hsa色氨酸残基的同步荧光最大吸收峰强度逐渐下降,表明药物与hsa形成了荧光较弱或无荧光的复合物。色氨酸残基的特征荧光光谱峰均能被药物猝灭,但hsa色氨酸荧光峰的位置变化不明显,表明药物对蛋白质分子构象的改变不大。而组合物(300k),随药物浓度的增加,hsa的同步荧光最大吸收峰强度逐渐上升,表明此温度下组合物可以增强同步荧光最大吸收峰强度,但hsa色氨酸残基的特征荧光光谱的峰位基本不变,表明组合物对蛋白质分子构象的改变不大。
7.3.3与has相互作用猝灭类型的确定
hsa的荧光猝灭机制包括静态猝灭和动态猝灭,猝灭机制一般由stern-volmer方程确定:
f0/f=1+kqτ0[q]=1+ksv[q](1)
kq=ksv/τ0(2)
式中:f0为无药物时,hsa的荧光发射强度,f为加入不同体积样品溶液后hsa的荧光强度,ksv为动态猝灭常数,kq为双分子猝灭过程速率常数,τ0为没有猝灭剂存在下生物大分子平均寿命(约为10-8s),[q]为样品溶液的浓度。
hsa溶液随活性单体和组合物溶液浓度增加对hsa的荧光猝灭的stern-volmer曲线见图20-24。
以f0/f对样品溶液浓度作图,得到样品溶液与hsa相互作用的stern-volmer曲线,求得速率常数及猝灭常数,结果见表3。
表3.温度为300k和310k时样品溶液与hsa的kq、ksv
猝灭剂对生物大分子的最大扩散碰撞猝灭速度常数一般是2.0×1010l/(mols)。根据表3由stern-volmer曲线求出的猝灭速度常数,在300k、310k时,样品溶液和hsa相互作用的猝灭常数kq的绝对值,均远大于2.0×1010l/(mols),则可以判断此猝灭过程均为形成无荧光复合物静态猝灭过程。
药物分子进入生物体后,在体内进行着吸收、代谢、确定靶器官和受体等重要的生理过程,而这些都是以大分子蛋白与小分子之间相互作用为桥梁进行的,所以蛋白与药物分子之间相互作用的研究为揭示药物在生物体内的作用机制具有重要作用。该研究可以为未来发现抗食管癌活性单体和组合物提供实验和理论基础,以及为通过合理设计血清白蛋白载体开发抗食管癌药物的靶向性治疗提供依据。
- 还没有人留言评论。精彩留言会获得点赞!