基于第三代测序的多倍体基因组组装方法和装置与流程
1.本发明涉及基因组组装技术领域,尤其涉及一种基于第三代测序的多倍体基因组组装方法和装置。
背景技术:
2.随着第三代单分子实时测序技术的发展,三代测序技术在基因组领域的应用已越来越广泛。简单基因组、重复基因组组装问题已经有了突破性进展,越来越多的简单基因组和高重复基因组已经组装出接近几百个间隔(gap)水平的染色体图谱。但是,在组装领域依然存在一些很复杂的基因组尚未成功获得基因组图谱,例如复杂四倍体的组装问题。
3.因此,利用三代单分子测序技术攻克更为复杂基因组的组装问题,成为近年来研究的一个热点。现有的三代组装软件(例如,mecat、canu、falcon、wtdbg等)主要基于二倍体基因组开发,对于组装比较纯合的异源多倍体表现出比较好的效果,目前已经发表的多倍体组装相关文章主要是关于异源多倍体的组装。但是复杂多倍体的组装目前还处于待解决状态,这是由于复杂多倍体染色体组之间的杂合性,以及多倍型带来的多重拷贝。现有的组装软件处理这种类型的情况普遍存在组装序列长度偏短、组装序列总长度远大于预估的基因组大小等问题,这往往导致后期挂载染色体困难,对生物学相关分析带来很大的干扰。
4.使用现有的二倍体组装软件组装复杂四倍体,获得的组装结果通常比预估的单倍体型的基因组大小的总长度偏大,但是无法确定组装结果是否包含一套完整的染色体组。在不清楚组装出基因组组成的情况下,往往不能很准确的筛选合适的方法通过去冗余来获得比较完善的基因组结果。因为现有的去冗余软件都是基于二倍体基因组开发的,主要利用基因组序列内部的相似性鉴定复杂序列进行去冗余处理,常用的去冗余软件有redundans、purge haplotigs、haplomerger2等。
5.hi-c(high-through chromosome conformation capture,高通量染色体构象捕获)技术,是染色体构象捕获(chromosome conformation capture,简称为3c)的一种衍生技术,是指以整个细胞核为研究对象,利用高通量测序技术,结合生物信息学方法,对全基因组范围内整个染色质dna在空间位置上的关系进行研究,通过对染色质内全部dna相互作用模式进行捕获,获得高分辨率的染色质三维结构信息。利用这种技术可以辅助基因组组装,并且通过同一条染色体的构象信息,获得整个基因组的染色体图谱。hi-c辅助组装主要是基于染色质片段间的交互强度呈现出随距离衰减的规律,根据hi-c测序读长(reads)的覆盖密度,判断组装序列的分类及相邻关系。目前hi-c辅助组装有lachesis、hirise、juicer+3d-dna三种软件,它们分别对基因组序列进行群组的划分、排序和定向,并对组装结果进行评估。由于hi-c的连接过程都是直接将hi-c的测序reads比对到原始的组装序列上,基于reads的覆盖情况来界定序列之间的相互关系,因此当需要连接的基因组序列内部存在很大的重复或者冗余,往往会导致相互关系界定异常,导致连接出现错误。
技术实现要素:
6.本发明的目的在于提供一种基于第三代测序的多倍体基因组组装方法和装置,有效地从复杂多倍体中分离出单套染色体组。
7.根据本发明的第一方面,本发明提供一种基于第三代测序的多倍体基因组组装方法,包括:
8.步骤1:获取多倍体基因组的三代单分子测序数据,并对其进行数据纠错和组装,得到基因组的第一组装结果;
9.步骤2:将三代单分子测序数据比对到第一组装结果进行深度评估并统计测序数据对整个基因组的覆盖度,获得组装出单拷贝和多拷贝的区域;
10.步骤3:选取组装出多拷贝的区域的序列,对其进行序列之间的比对以去除覆盖在多拷贝区域内的序列之间的重复,得到第一轮去冗余结果;
11.步骤4:对第一轮去冗余结果,鉴定并打断可能的错误连接后对基因组序列重新拼接以去除基因组上的拼接问题,得到接近预估的单套染色体基因组大小的第二组装结果;
12.步骤5:对第二组装结果,判断保守基因数及多拷贝保守同源基因数的变化情况以确定去冗余成功;并且,将第一组装结果中未包含到第二组装结果的部分序列合并到第二组装结果,然后对组装结果进行优化和矫正,得到第三组装结果。
13.在优选实施例中,所述多倍体是四倍体。
14.在优选实施例中,所述步骤2中使用纠错后的三代单分子测序数据比对到第一组装结果进行深度评估并统计测序数据对整个基因组的覆盖度。
15.在优选实施例中,所述步骤3包括:首先,从组装出多拷贝的区域的序列中选取最优比对的序列标记为候选的多拷贝序列,然后,对候选的多拷贝序列再次比对筛选。
16.在优选实施例中,对候选的多拷贝序列再次比对筛选的步骤进行多轮的迭代,以防止折叠重复和嵌合序列的干扰,确保最终得到的单拷贝序列不再与其他序列存在多重拷贝关系,从而得到所述第一轮去冗余结果。
17.在优选实施例中,所述步骤4中的重新拼接过程中,若两个等位基因共享同一位点,则将两个等位基因分别组装到两个独立的单倍型的组装子序列结果中;若一个等位基因只对应一个位点,则将该位点分别放在两个独立的单倍型的组装子序列结果中,并尽可能保证一套组装子序列包含完整基因组拼接序列。
18.在优选实施例中,所述方法还包括:
19.步骤6:对第三组装结果进行hi-c连接,得到第三组装结果的hi-c连接结果,以便对第三组装结果进行校验和评估。
20.在优选实施例中,所述方法还包括如下步骤7至9中至少一个步骤:
21.步骤7:将第三组装结果的hi-c连接结果与第一组装结果进行序列比对,验证第三组装结果的完整性和第一组装结果的序列组成和成分。
22.步骤8:通过比较第一组装结果和第三组装结果的hi-c连接结果的完整保守基因数目,预估第三组装结果的完整性。
23.步骤9:将三代单分子测序数据比对到第三组装结果的hi-c连接结果中,验证三代单分子测序数据的利用率和在整个基因组水平的覆盖情况。
24.在优选实施例中,所述步骤7包括:
25.选择特异性限制性内切酶,利用模拟酶切将第一组装结果和第三组装结果的hi-c连接结果分别转化为代表酶切位点位置的数据格式,进行酶切位点的比对,得到第一组装结果和所述hi-c连接结果之间的相互关系,进而确定组装方法的可靠性。
26.根据本发明的第二方面,本发明提供一种基于第三代测序的多倍体基因组组装装置,包括:
27.纠错和组装单元,用于获取多倍体基因组的三代单分子测序数据,并对其进行数据纠错和组装,得到基因组的第一组装结果;
28.比对和校验单元,用于将三代单分子测序数据比对到第一组装结果进行深度评估并统计测序数据对整个基因组的覆盖度,获得组装出单拷贝和多拷贝的区域;
29.结果去重复单元,用于选取组装出多拷贝的区域的序列,对其进行序列之间的比对以去除覆盖在多拷贝区域内的序列之间的重复,得到第一轮去冗余结果;
30.结果重组单元,用于对第一轮去冗余结果,鉴定并打断可能的错误连接后对基因组序列重新拼接以去除基因组上的拼接问题,得到接近预估的单套染色体基因组大小的第二组装结果;
31.校验和优化单元,用于对第二组装结果,判断保守基因数及多拷贝保守同源基因数的变化情况以确定去冗余成功;并且,将第一组装结果中未包含到第二组装结果的部分序列合并到第二组装结果,然后对组装结果进行优化和矫正,得到第三组装结果。
32.根据本发明的第三方面,本发明提供一种计算机可读存储介质,其包括程序,上述程序能够被处理器执行以实现如第一方面的方法。
33.本发明主要通过三代单分子测序数据对复杂多倍体进行拼接,并结合三代测序技术和hi-c技术方法,对复杂多倍体原始组装结果进行校验,基于校验结果优化复杂多倍体的组装结果及相应的去冗余处理,最终从复杂的多倍体组装结果中获得单套染色体组型,解决了复杂多倍体组装的一个重要的技术难题。同时,优选实施例还提供基于酶切的基因组序列之间的快速比对和校验方法,和鉴定单倍体染色体组上的同源、异源、杂合的区域,为后续的多倍体染色体整体组装和染色体分型提供技术依据。
34.具体而言,本发明通过三代测序数据对原始组装结果的覆盖情况,初步推断出复杂多倍体原始组装结果的主要构成和基因组的复杂情况。通过深度覆盖情况对组装的原始组装结果的结构进行分析,确定多倍体基因组组装的基本方案。如果组装出多套完整的染色体组对应的序列,则通过技术手段分离出完整的几套染色体组。如果组装出的基因组只包含有一套完整的染色体组对应的序列,但是可能由于序列复杂性,有部分区域组装出其他几套染色体对应的序列,则需要利用技术手段,例如去冗余处理,从原始组装结果中成功分离出只包含完整的一套染色体组序列。这样就可以很大程度上简化多倍体基因组的组装。
35.本发明提供了有效去除冗余的方法。首先,结合三代测序数据基于基因组的覆盖度,对于可能存在组装异常的序列进行过滤,再对可能的重复区域进行序列之间的相似性鉴定和序列分型;然后,在第一轮去冗余的基础上,再通过全基因组水平的序列相似性比对去冗余。两轮嵌套的去冗余方法,相较于只使用其中之一的方法,能够有效去除基因组内部的重复。通过全基因组的保守同源基因数目的评估,重复基因的数目有明显水平的降低,但是全基因组水平的保守基因数目变化很小。通过对去冗余后的结果再进行多种组装方法的
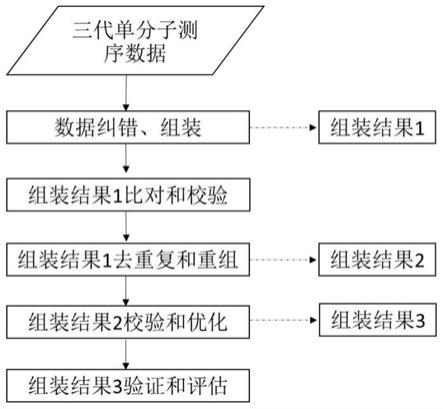
合并和序列添加,contign50指标能够明显提升。并且去冗余的结果进行hi-c连接,得到的染色体分布更为均匀,更接近真实染色体的大小。
36.本发明提供了组装结果的校验方法。通过对hi-c连接结果与原始组装结果通过模拟酶切的方法进行比较,快速比对出原始的组装结果对整个多倍体基因组的整体分布情况。同时结合三代测序数据对整个染色体水平的覆盖度,推测最终组装结果中包含的同源区域及异源区域,为后续基因分型和全套多倍体的组装提供技术依据。
37.概言之,本发明的方法能够有效地从复杂多倍体中分离出单套染色体组,为获得复杂多倍体的其他染色体组的组装奠定基础,同时对处理高重复高杂合的基因组组装也提供了很好的技术依据,该方法在基因组组装领域有广阔的应用前景。
附图说明
38.图1为本发明实施例中基于第三代测序的多倍体基因组组装方法流程图。
39.图2为本发明实施例中不同深度覆盖对应的组装结果情况,其中s1为组装结果为4个拷贝的情况;s2为组装结果为2个拷贝的情况;s3为组装结果为1个拷贝的情况;s4为多个拷贝混合的情况。
40.图3为本发明实施例中实际组装结果之间通过酶切的方法比对,并利用可视化界面展示的具体情况,其中302为1个拷贝,304为2个拷贝,306为多个拷贝混合的情况。
41.图4为本发明实施例中测试物种的深度分布情况及分析过程,其中s4为实际的深度分布图,s1+s2+s3为猜想的一种组装结果的组合形式。
42.图5为本发明实施例中测试物种最终组装结果的hi-c连接结果和原始组装结果之间的比对情况,存在很多的二重拷贝、三重拷贝的区域。
43.图6为本发明实施例中最终组装结果的深度分布情况,组装结果3的染色体连接结果中,主峰在21和86左右的位置,但是在中间有较高的跨度,推测可能该四倍体比较复杂,单倍型中同时存在1个拷贝、2个拷贝、3个拷贝和4个拷贝的区域。
具体实施方式
44.下面通过具体实施方式结合附图对本发明作进一步详细说明。在以下的实施方式中,很多细节描述是为了使得本发明能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他材料、方法所替代。
45.另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
46.本发明中,三代单分子测序包括但不限于pacbio或者nanopore的测序方式。
47.本发明中,组装结果1也称作“第一组装结果”,组装结果2也称作“第二组装结果”,组装结果3也称作“第三组装结果”。
48.本发明以下实施例中,以四倍体的组装为例来说明基于第三代测序的多倍体基因组组装方法,其他多倍体的组装原理和方法与此相同,使用本发明的方法可解决高杂合高重复的二倍体或者其他复杂多倍体的组装。
49.本发明一个实施例中,提供一种复杂四倍体的组装方法,具体过程如图1所示,包括:
50.a)三代序列纠错及组装。主要基于三代单分子测序数据进行数据纠错和组装,包括对全基因组测序数据进行数据纠错,纠错后数据拼接,拼接结果分型和拼接结果抛光处理得到高精确度的初级基因组的组装结果1。常用的三代相关分析软件包括falcon、mecat、canu、wtdbg等。
51.b)组装结果1比对和校验。用三代单分子测序数据直接比对到初级组装结果1进行深度评估,并统计三代单分子测序数据对整个基因组水平的覆盖度。此处单分子测序数据可以是原始未纠错的数据,也可以是经过纠错后的数据。理论上讲,为了提高准确性,推荐使用纠错后的数据进行覆盖度统计分析。比对软件可以是任何可以进行三代长reads数据比对的软件,例如bwa、blasr、minimap2等,比对文件的输出格式为bam格式,可以使用samtools或者bedtools软件进行深度统计。如果相对于四倍体的单套染色体的测序深度为4n,通常四倍体基因组常见的深度覆盖情况包括三种,如图2:
52.(1)当所有区域都组装出1个拷贝,理论上讲,组装结果应该接近于单套染色体长度的总和,并且每个位点都应该有测序数据覆盖,所有位点的覆盖近似服从泊松分布,覆盖的峰值应该在平均测序深度4n的位置附近,如图2中s3。
53.(2)当所有的区域都组装出2个拷贝,理论上讲,组装结果应该接近于单套染色体长度的2倍,深度分布的峰值应该在2n的位置附近,如图2中s2。
54.(3)当所有区域都组装出4个拷贝,理论上讲,组装结果总大小应该接近于单套染色体长度的4倍,并且深度分布的峰值应该在n的位置附近,如图2中s1。
55.但是对于一些复杂四倍体,总长度不是单倍、二倍、四倍的单倍型预估基因组大小,则可能在一些区域组装出多个拷贝,表现在深度如图2中的s4的样子。如果是图2中s4的峰型,则组装结果较为复杂,同时包含多种类型的组装拷贝区域,如果从原始组装结果直接获得完整四套复杂染色体,由于序列之间的相似性、杂合性和异源性,染色体直接分型方面在技术实现上较为困难。但是从原始组装结果中首先保留单套完整的染色体,然后再通过其他技术手段基于单套染色体进行分型处理,从技术实现上则相对容易。因此本发明的方法主要提供从原始组装结果中分离单套染色体进行组装的方法。
56.c)组装结果1去重复和重组。现有的二倍体去冗余软件进行去冗余,无法处理多个倍型导致的冗余问题,测试发现多个去冗余软件的测试显示最终结果仍然包括大于1倍的单套染色体基因组大小。因此,考虑采用不同的去冗余方法嵌套使用。具体方法如下:
57.(1)结果去重复。基于三代测序数据对原始组装结果1的覆盖情况,可以确定深度分布的最低点(如图2中s4对应的a点)、拐点(如图2中s4对应的b点和c点)、最高点位置(如图2中s4对应的d点)。当覆盖深度接近单倍型基因组大小的覆盖深度在图2中s4上[c,d]区域内,说明该区域组装出单个拷贝,而如果单倍型基因组大小的覆盖深度为区域覆盖深度在图2中s4上[a,c]区域,说明该区域组装出多个拷贝。选取组装出多个拷贝的区域序列,即s4上[a,c]区域内的序列,只对这个区域内的序列进行序列之间的比对和杂合位点的鉴定。首先,利用blast比对软件选取最优比对的序列标记为候选的多拷贝序列;然后,利用lastz软件对候选的多拷贝序列再次比对筛选。为防止折叠重复和嵌合序列的干扰,lastz软件比对将进行多轮的迭代,确保最终得到的单拷贝序列不会再与其他序列是多重拷贝关系,从
而得到最终的组装结果,称为第一轮去冗余结果。特别强调的是,在这个过程中,深度覆盖过低(小于图2中s4的a点)或者过高(超过图2中s4的d点)的区域,如果组装序列长度的绝大部分碱基(例如70%以上的碱基覆盖)在该区域,则认为这些组装序列是无效的组装序列,在分析过程中这部分序列被直接丢弃。具体序列丢失的问题,会在后续分析中不断补充进去。第一轮去冗余过程的实现使用二倍体去冗余软件purge haplotigs:https://www.biorxiv.org/content/early/2018/03/22/286252。使用purge haplotigs的去冗余结果相对保守,它只针对深度覆盖存在多重拷贝的区域进行比对和处理,而且只是将可能组装出重复的序列去掉,序列本身不会发生变化,因此用在第一步去冗余操作比较合适。
[0058]
(2)结果重组。基于第一轮去冗余的结果,已经去除掉了大部分深度覆盖在多重拷贝区域内的序列之间的重复。因此基于第一轮去冗余结果,可以把组装结果简化为类似于二倍体重复与杂合位点鉴定及分离的问题,而去冗余软件haplomerger2可以实现序列之间的重新装配和重组。这是基于haplomerger2是利用全基因组水平的序列比对迭代,鉴定并打断可能的错误连接,再利用比对结果进行基因组序列的重新拼接。这个拼接过程中,如果两个等位基因共享同一位点,则将对应的两个等位基因分别组装到两个独立的单倍型的组装子序列结果中;如果一个等位基因只对应一个位点,则将这个位点分别放在两个独立的单倍型的组装子序列结果中,并且尽可能保证一套组装子序列包含完整基因组拼接序列。通过这种方法,就可以有效去除基因组上由于多重拷贝和重复嵌套导致的拼接问题,从而得到接近预估的单套染色体基因组大小的组装序列,称为组装结果2。序列打断和重组使用的二倍体去冗余软件haplomerger2可从https://github.com/mapleforest/haplomerger2/releases/获得。
[0059]
d)组装结果2的校验和优化。利用busco评估(与保守的单拷贝的直系同源基因集的完整性评估https://busco.ezlab.org/),判断保守基因数目以及多重拷贝的保守同源基因数目变化情况。如果去冗余成功,保守基因的数目变化应该不会很大,但是多重拷贝的保守同源基因数目会明显降低。此外,为了保证整个去冗余之后,不会导致整个基因组序列信息的丢失,我们采用quickmerge(https://github.com/mahulchak/quickmerge)软件,基于mummer的比对结果将组装结果1中未包含到组装结果2的部分序列与组装结果2的结果合并,将可能存在重叠(overlap)的区域再次进行补充和添加。再利用haplomerger2软件,基于序列内部比对,将可能的错误连接打断、单倍型序列重组、手工添加的方式丢失的序列等方法对组装结果进行优化,然后通过三代测序数据利用抛光软件对优化后的序列进行矫正,得到组装结果3。组装结果3更接近预估的单套染色体大小,而且通过不断去重复拷贝之后的序列拼接,可以使序列的contig n50有很大程度的提升,甚至达到1m以上的水平。利用组装结果3进行hi-c连接,使用的hi-c数据是基于组装结果1过滤之后的hi-c数据。直接用组装结果1进行hi-c连接,得到的染色体长度偏差很大,尤其部分染色体长度达到几倍的差异,整个染色体大小也是多个拷贝的基因组大小;而利用组装结果3进行hi-c连接,连接结果基本接近单拷贝的基因组大小,并且染色体长度分布基本相近或者相差不大。
[0060]
e)组装结果3的校验和评估。
[0061]
(1)模拟酶切的方法进行结构校验。利用组装结果3的hi-c连接结果和组装结果1进行序列比对,验证组装结果3的完整性和组装结果1的序列组成和成分,如果基于传统的比对方法比对时间久,比对速度慢,而且当组装结果序列较多时,比对结果无法直观地将序
列的共线相关性展示出来。本发明提供一种基于模拟酶切的方法进行基因组之间的快速比对和校验。基于bionano分子图谱比对速度快的特点,在组装结果上利用模拟酶切的方法,选择合适的特异性的限制性内切酶将基因组序列的fasta格式转化为代表酶切位点位置的cmap格式,这样利用酶切位点之间的比对相对于传统的比对有更快的速度和更低内存的消耗。比对软件使用bionano分子图谱的比对软件refalign,可以从bionano solve软件包(https://bionanogenomics.com/support/software-downloads/)中获得。由于refalign的软件要求,需要保证整个基因组范围内每100k范围内酶切位点的分布密度约为8-25左右。利用设计好的酶,将组装结果1和组装结果3的hi-c连接结果分别转化为cmap格式,进行酶切位点的比对。利用这种方法虽然比对精度不高,但是可以利用bionano分子比对的可视化界面清晰看到原始组装结果和最终组装结果之间的相互关系,从而进一步确定组装方法的可靠性。如果通过hi-c把单套染色体的序列连接成为染色体,可以更准确的看到每条染色体上原始序列的大致分布情况。校验结果类型如图3,其中,302显示原始组装结果相对于hi-c的染色体序列只有1个拷贝;304显示原始组装结果相对于hi-c的染色体图谱有2个拷贝;306显示原始组装结果相对于hi-c的染色体序列部分区域为1个拷贝,部分区域为2个拷贝,部分区域为3个拷贝,部分区域为4个拷贝,306显示序列之间的比对情况较为复杂,由于部分区域序列差异导致酶切位点不一致,从而导致该区段组装出4个拷贝的序列。通过这种方式,可以更进一步确定原始组装结果的均一性和验证最初通过深度分布验证组装结果的可能组成的正确性。
[0062]
(2)基于保守的单拷贝直系同源基因的完整性校验(busco评估)。通过比较组装结果1和组装结果3的hi-c连接结果完整的保守基因数目,可以预估最终组装出来的序列是否完整。如果最终获得的单拷贝直系同源基因包含完整的一套单倍型染色体,保守基因总数目应该变化不大,但是单拷贝的保守基因所占的比例应该会增加,而多拷贝的保守基因的比例会明显下降。
[0063]
(3)基于覆盖深度评估。将三代单分子测序数据比对到组装结果3的hi-c连接结果中,验证三代单分子测序数据的利用率和在整个基因组水平的覆盖情况,类似于图2的方法,在去冗余之后,通过深度覆盖情况,可以更清晰地区分整个基因组区域的单倍型大小覆盖深度区域、二倍型大小覆盖深度区域、四倍型大小的深度覆盖区域,基于统计计算区域面积比例,大致可以确定整个基因组上不同倍型的染色体区域的分布。从而为同源性和异源性区域判断,基因组分型和全套染色体序列的获得奠定基础。
[0064]
以下通过实施例详细说明本发明的技术方案和技术效果,应当理解实施例仅是示例性的,不能理解为对本发明保护范围的限制。
[0065]
以下实施例是一个具体的基因组组装实施例。基因组为一种四倍体植物,单倍型基因组大小预估约为293mb。在此实施例中,基于三代pacbio测序数据和hi-c数据对基因组进行组装,使用的三代数据为48g,相对于单倍型基因组大小约164x的覆盖深度;hi-c数据140g,约478x。具体方法操作步骤如下:
[0066]
1.三代序列纠错及组装。使用三代组装软件falcon(v0.7)对三代pacbio的测序数据进行纠错和组装,并使用falcon-unzip尝试对原始组装结果进行分型处理,并使用smartlink官方的基于三代pacbio数据的polish分析软件arrow进行组装结果矫正,得到组装结果1。其中组装结果1的序列总长度为726m,n50长度为296k,大于500bp的序列总条数为
3834条。原始组装结果1的726m比预估的单倍型基因组大小的293m的2倍还要大,要完成该四倍体的基因组组装,需要先了解726m的序列之间的相互关系。
[0067]
2.组装结果比对和校验。用三代pacbio数据的falcon纠错结果(25g,约85x的单倍型覆盖深度)直接比对到组装结果1,并统计三代单分子测序数据对整个基因组水平的覆盖度,根据深度验证组装结果序列之间的相互关系。三代软件使用bwa(v0.7.12-r1039)进行序列比对,并用samtools(v1.3)进行数据统计。最终比对上参考序列的数据量有22.5g,深度分布情况如图4的s4峰值,主峰值位置在21x的位置,而且主峰后面有较大的拖尾,并不是标准的泊松分布图的曲线形式,因此预估组装结果1中包含大量的多重拷贝,而二重拷贝和单拷贝的序列占得比重较少,而四重拷贝的数目过多导致其他拷贝的序列深度分布不明显,图4列出一种最可能的峰值组合,即s1、s2、s3的组合方式。因为用22.5g的数据量,左侧深度分布峰值为21,在标准泊松分布下,预估基因组大小应该为1.07g,但实际组装结果只有726m,还应存在基因组大小约为345m导致峰图不服从泊松分布,而是有个很长的拖尾。所以组装的726m的组装结果1并不是4倍的单倍型基因组大小,也不是完整的四套染色体。因此如果从组装结果1直接复原完整四套复杂染色体,在技术实现上相对较为困难。比较简单和高效的办法是将混合组装结果只保留单套染色体,然后再通过其他技术手段对单套染色体进行分型处理,从而得到完整的四套染色体。
[0068]
3.组装序列的去重复和重组。首先使用二倍体去冗余软件purge_haplotigs(https://bitbucket.org/mroachawri/purge_haplotigs)进行组装结果1的冗余去除,得到第一轮去冗余结果。purge_haplotigs在去冗余的过程中,首先把深度过高和过低的序列当作无效序列被去除掉,再从保留的区域中,鉴定出组装出多个拷贝的区域和多倍体只装出单倍型的区域。因为深度分布过低的序列可能为不可信序列,而深度分布过高的序列可能来自于细胞器的区域,所以这些序列在最初鉴定重复时作为无效序列被过滤掉。根据图4中的分析,选取a=3,c=60,d=120作为深度的最低点,多拷贝和单拷贝的临界点和深度的最高点,在序列有70%以上的序列都在[3,120]区间外的序列都被过滤掉。对深度分布[3,60]范围内的序列进行去冗余,得到第一轮去冗余结果。
[0069]
结果显示,第一轮的去冗余结果依然有超过100m的可能组装出多个拷贝的序列,因此在第一轮去冗余结果的基础上,利用去冗余软件haplomerge2进行去冗余处理,一方面可以使组装出的拷贝数目大大降低,更接近与二倍体水平进行去冗余操作,另一方面也可以有效避免全基因组范围内序列相似性造成的干扰。haplomerge2的分析参数选取系统给的的默认参数。haplomerge2可以将可能存在连接冲突的组装序列打断,再基于比对结果将共享等位基因位点分开成为二套单倍型组装结果,并在分析过程中尽可能保证一套单倍型结果的完整性和准确性,作为主要组装结果。通过haplomerge2处理之后的主要结果序列总长度为356m,基本接近预估的单倍型的基因组大小,称为组装结果2。本发明也测试了直接使用两种去冗余软件进行序列去冗余,purge_haplotigs得到的基因组大小为531m,haplomerge2得到的基因组大小为463m,最终都远超1个拷贝的基因组大小,所以两种方法合并使用可以去掉更多的重复。
[0070]
4.序列的校验和优化。虽然组装结果2的结果更接近于基因组大小,但是经过两轮去冗余处理,依然还是比预估单倍型基因组大小偏大50m左右,这可能因为原来组装序列中,由于四倍体中存在重复和杂合位点的干扰,导致一些区域组装存在拼接重复。因此为了
保证能把这些区域完整分离出来,利用组装结果1对组装结果2进行补充,将有重叠但不相同的序列补充到组装结果2,再进行序列的打断和重排,可能更有机会拼接错误导致的重复。将组装结果1中未包含在组装结果2的序列分离出来,采用quickmerge软件,基于mummer的比对分析,对组装结果2的序列进行补充和添加。因为添加后的序列,还可能引入冗余的序列,因此再次使用haplomerge2软件,对序列进行打断和重新组装,最后通过三代pacbio原始测序数据基于pacbio官方的纠错软件arrow对序列进行矫正。最终得到contig n50为1.5m,总长度为300m的组装结果,这个结果接近于预估的单倍型基因组大小293m,称为组装结果3。对组装结果3进行hi-c连接,直接分出1套完整的单倍体型基因组大小的染色体,与直接用组装结果1进行hi-c连接相比,得到的染色体长度更为均匀,更接近真实的染色体长度。总体的分析结果的比较如表1。
[0071]
表1
[0072][0073]
5.基于酶切的方法对组装结果3进行快速比对校验。用模拟酶切的方法选择特异性的酶bsqqi,在整个基因组内部该酶切位点的分布密度为12个酶切位点/100k。利用设计好的酶,将组装结果1和组装结果3的hi-c连接结果转化为cmap格式,然后利用refalign软件进行酶切位点之间的比对。将比对结果导入到bionano的可视化分析软件irysview中,可以快速观测到组装结果1和组装结果3的结构关系。图5示出了组装结果3的hi-c连接结果中某一条染色体的酶切位点的比对情况。最上面一排连续的是hi-c连接后的组装结果3,而下面三排比较短的每一块,代表组装结果1的一条序列,图中过滤掉confidence(可视化界面的参数)小于20的比对结果。从图中可以看到正如图4预估出的样子,部分区域是1个拷贝,还有很大一部分区域有超过2个拷贝,4个拷贝的序列占的比例较少。其他染色体的情况与该染色体类似,不在此处全部列出。
[0074]
6.基于保守的单拷贝直系同源基因的完整性校验(busco评估)结果如表2。表2示出整个过程中组装指标和busco评估结果的变化,其中busco评估选取的模型embryophyta_odb9,选取的augustus训练物种是arabidopsis。最终结果显示总的保守基因占得比例相差
不大,但是多拷贝保守基因的比例明显下降。
[0075]
表2
[0076][0077]
7.组装结果3的评估。将falcon的纠错结果比对到组装结果3的hi-c连接结果中,验证最终结果的三代数据覆盖情况,结果如图6。图6显示组装结果3的染色体连接结果中,主峰在21和86左右的位置,但是在中间有较高的跨度,推测可能该四倍体比较复杂,单倍型中同时存在1个拷贝、2个拷贝、3个拷贝和4个拷贝的区域。而且存在单拷贝和四个拷贝的区域,可以根据深度分布把它们分离出来,具体2个拷贝和3个拷贝的信息,需要借助其他测序手段进行分离,从而完成四倍体全套染色体的组装。
[0078]
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1