一种肝细胞肝癌患者诊断、预后和复发统一模型的构建方法与流程
本发明涉及生物领域,尤其涉及一种肝细胞肝癌(hcc)诊断、预后和复发统一模型的构建方法。
背景技术:
肝细胞肝癌(hcc)是肝癌的最常见类型,癌症相关死亡的第三大原因,以及全球主要的侵袭性的恶性肿瘤。肝癌是一个多步骤且复杂的疾病,涉及一系列遗传和表观遗传学改变,包括基因组缺失,扩增,突变和/或插入。早期诊断和介入治疗,以及治疗方法和手术方法的发展,已推进治疗该癌症的重大进展。然而,绝大多数的晚期肝癌患者确诊时预后不良。因此,迫切需要更好地了解hcc的功能通路和分子机制,以及开发可用于早期诊断和预测预后与复发的关键新型生物标志物。
表观遗传的改变被普遍认为可影响基因表达,dna甲基化,非编码dna和组蛋白乙酰化等遗传修饰。dna甲基化是一种主要的表观遗传修饰,参与基因的转录调控并保持基因组的稳定性。各种癌症具有特殊的调节紊乱,其特征在于异常的dna甲基化,它调节了许多肿瘤相关基因的表达,对肿瘤的发展至关重要。甲基化的变化,其中包括原癌基因的低甲基化和肿瘤抑制基因的高甲基化,这些被认为是在包括hcc在内的癌症发生关键事件。因此,检测dna甲基化驱动基因并了解与这些基因相关的分子作用机制可能有助于阐明hcc的发病机理和致病机制。近年来,一些使用全基因组测序检测癌症的甲基化数据表明,大量的基因在肿瘤中表现出异常的dna甲基化。此外,这些变化可以用于癌症亚型分类和预测癌症预后。总体而言,鉴定在癌症的发生发展持续过程中通过dna甲基化介导的基因沉默作用来充当“驱动基因”的基因以及仅在致癌过程中充当“乘客基因”的那些基因可能有利于发展最佳靶向表观遗传学治疗方法。然而,由于人类癌症中存在许多甲基化差异的基因,因此筛选出明显的驱动基因和乘客基因非常困难。
hcc发病机理是一个复杂的生物学过程,涉及遗传因素和表观遗传学的变化,而dna甲基化升高是hcc发展的早期事件。一项荟萃分析提供了经验证据,表明细胞因子信号传导抑制因子1的异常启动子甲基化可能导致hcc的发生。既往研究报道,在表观遗传学上,酒精相关性肝癌的视黄醇代谢的基因和丝氨酸羟甲基转移酶1通过启动子dna甲基化受到调节。然而,大多数研究主要集中在甲基化或基因表达数据上,并且没有进行联合分析。因此,对驱动hcc的细胞和分子机制缺乏全面的了解,从而限制了治疗策略。
技术实现要素:
有鉴于此,本发明的目的之一是提供一种肝细胞肝癌患者诊断、预后和复发的统一模型,该模型可以有助于临床对肝细胞肝癌的治疗指导。
本发明通过以下技术手段解决上述技术问题:
一种肝细胞肝癌患者诊断、预后和复发统一模型的构建方法,包括以下步骤:
step1)采集hcc的基因表达数据和dna甲基化数据;其中,采集hcc的基因表达数据过程中需要对hcc中差异基因进行筛选,其步骤如下:从tcga获得hcc组织(n=371)和非肿瘤组织(n=50)中的mrna表达谱。使用fdr<0.05和|log2fc|>1作为筛选差异表达基因(deg)的阈值,满足条件的总共有9,219个deg,其中表达上调的有7,734个deg,表达下调的有1,485个deg,均选择用于后续分析。
step2)对hcc的基因表达数据和dna甲基化数据进行分析,鉴定出hcc的dna甲基化驱动基因;具体说来,为了鉴定hcc中的dna甲基化驱动基因,纳入了包括tcga中792个临床样品中9,219个deg的基因表达和dna甲基化数据(371个hcc和50个非肿瘤样品的dna甲基化数据以及371个hcc样品的配对基因表达数据)。在methylmix分析中,总共筛选出了123个dna甲基化驱动的基因。在这些基因中,有77个是高甲基化基因、46个是低甲基化基因。差异甲基化基因的筛选标准为fdr<0.05,dna甲基化与基因表达之间的相关性<-0.3。然后,我们使用365个hcc样本以及生存时间和生存状态,使用单因素cox回归分析研究了123个dna甲基化驱动基因的表达与预后之间的关系。在123个dna甲基化驱动基因中,有51个具有统计学意义(p<0.05)。随后使用lasso方法进行分析,lasso是一种惩罚性回归方法,使用l1罚分将回归系数缩小为零,从而基于以下原则消除了许多变量:当罚分较大时,选择的预测变量变少。因此,具有非零系数的种子基因被认为是潜在的预后指标。基于r语言的glmnet包使用1000次coxlasso回归迭代和10倍交叉验证,将种子基因缩小为多基因集。具有非零系数的基因被认为是潜在的预后基因。在coxlasso回归的1000次迭代中出现的非零系数越高,该基因预测预后的能力就越强。通过coxlasso回归的1000次迭代分析了前期结果获得的51个dna甲基化驱动基因,以进一步减少数量筛选出重要的基因。
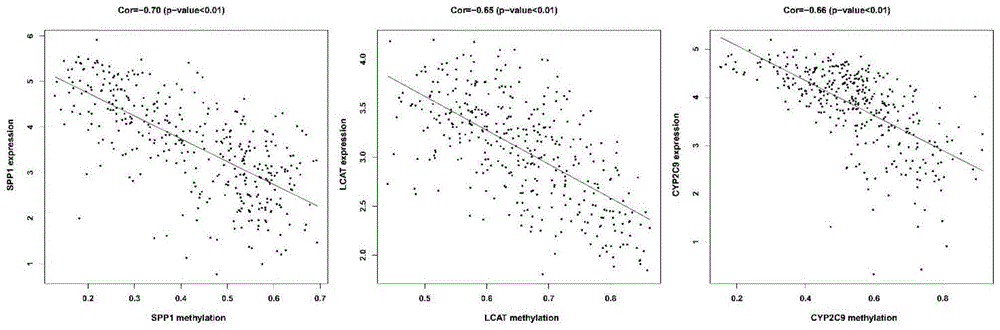
通过应用lasso分析后,结果鉴定了3个dna甲基化驱动的预后基因,分别是分泌磷蛋白1(secretedphosphoprotein1,spp1)、卵磷脂胆固醇酰基转移酶(lecithin-cholesterolacyltransferase,lcat)和细胞色素p450家族2亚家族c成员9(cytochromep450family2subfamilycmember9,cyp2c9)。相关性分析结果表明,spp1、lcat和cyp2c9的基因表达与dna甲基化状态具有显著的负相关性。
step3)优选的,在验证集中验证步骤2中所述的建立预后模型的具体方法如下:利用多因素cox比例风险模型的回归系数建立肝细胞肝癌患者诊断、预后和复发统一模型,所述预后-复发-诊断评分运用以下公式计算:
预后-复发-诊断评分=(0.29344×spp1基因表达水平)–(2.3052×lcat基因表达水平)–(0.21059×cyp2c9基因表达水平);
我们在训练集(tcga)中使用x-tile软件寻找最佳阈值。最佳阈值定义为在mantel-cox检验中产生最大χ2的风险评分。通过最佳阈值(1.32)将患者分为高风险和低风险患者。
在训练集中,与低风险患者相比,高风险患者的生存时间(危险比,hr=2.72,95%置信区间,95%ci=1.81-4.09,p<0.001)显著更差。根据上述的公式和阈值,我们在验证集(gse14520)中进一步测试了该模型预测预后的能力。与训练集的结果一致,验证集中的高风险患者的生存时间显著短于低风险患者(hr=1.64,95%ci=1.09-2.45,p=0.008)。
优选的,我们用上述的公式和阈值测试了肝细胞肝癌患者诊断、预后和复发统一模型预测肝细胞肝癌复发的能力。在训练集中,高风险患者的复发率(hr=1.82,95%ci=1.12-2.96,p=0.003)显著高于低风险患者。同时我们在验证集中评估了该模型预测复发的性能。与训练集的结果一致,高风险患者的复发率显著高于低风险患者(hr=1.54,95%ci=1.03-2.29,p=0.02)。
优选的,我们还表明,该模型能够在训练集(auc=0.981)和验证集(auc=0.952)中显著区分正常样本与肝细胞肝癌。
总之,这些结果证明了3个dna甲基化驱动基因的表达水平对于构建肝细胞肝癌患者的诊断,预后和复发统一模型具有较强的应用价值。
图1为预后-复发-诊断统一公式中spp1、lcat和cyp2c9甲基化与表达关系图,结果显示在hcc组织中这3个基因的表达水平与甲基化水平显著负相关,随着甲基化程度的升高,基因表达降低。预后-复发-诊断统一模型的预测肝细胞肝癌患者预后的结果,如图2所示,表明预后-复发-诊断评分高的肝细胞肝癌患者预后不良。在如图3所示的复发图中,预后-复发-诊断评分高的肝细胞肝癌患者复发率较高。如图4所示,表明预后-复发-诊断统一模型的诊断肝细胞肝癌性能优异。
本发明的有益效果:
本发明发现并验证了由3个dna甲基化驱动基因组成的肝细胞肝癌患者诊断、预后和复发统一模型。另外,通过整合多维基因组数据获得的结果为肝细胞肝癌生物标记物提供了新的研究方向,并为肝细胞肝癌患者的个体化治疗提供了新的可能性。
附图说明
下面结合附图和实施例对本发明作进一步描述。
图1为spp1、lcat和cyp2c9的dna甲基化与基因表达的关系图;
图2为肝细胞肝癌患者诊断、预后和复发统一模型在训练集(tcga)和验证集(gse14520)中预测肝细胞肝癌患者预后的示意图;
图3为肝细胞肝癌患者诊断、预后和复发统一模型在训练集(tcga)和验证集(gse14520)中预测肝细胞肝癌患者复发的示意图;
图4为肝细胞肝癌患者诊断、预后和复发统一模型在训练集(tcga)和验证集(gse14520)中诊断肝细胞肝癌的示意图。
具体实施方式
以下结合具体实验对本发明作详细的说明:
本发明:一种肝细胞肝癌患者诊断、预后和复发统一模型的构建方法,具体包括以下步骤:
步骤一:数据收集与分析
1)收集患者样本
从癌症基因组图谱(tcga)数据库中获得了总共421个rna测序数据(371个hcc样品和50个非肿瘤样品),430个dna甲基化数据(380个hcc样品和50个非肿瘤样品)以及相应的hcc患者临床信息。在380个hcc样本中可获得dna甲基化数据中,其中371个hcc样本既包含rna测序数据又包含配对的dna甲基化数据。在371个tcgahcc样本中,有365个包括总生存时间(os)和生存状态,而317例包括无病生存时间和复发状态。使用illuminahiseq2000rna测序平台从tcga数据库获得hcc基因表达数据,并使用illuminainfiniumhumanmethylation450平台获得dna甲基化数据。计算基因启动子,位于转录起始位点(tss)1500和tss200中所有cpg位点的平均dna甲基化值,作为该基因的dna甲基化值。gse14520微阵列数据集包括基因表达谱(225个hcc样本和220个非肿瘤样本)和相关的临床数据。在gse14520数据集中的225个hcc样本中,有221个包括总生存时间和生存状态,有221个包括无病生存时间和复发状态。
2)在肝癌中筛选差异表达基因(deg)
为了鉴定关键的hcc发展的基因,我们使用“edger”r软件包从tcga中鉴定了371个hcc样品和50个非肿瘤样品之间的deg。错误发现率(fdr)<0.05,|log2倍数变化(fc)|>1被用作阈值。
3)基因表达数据和dna甲基化数据的联合分析
使用r语言中的methylmix软件包用于分析371个hcc样品和50个非肿瘤样品的dna甲基化数据以及371个hcc样品配对的基因表达数据,以鉴定对相应基因表达有重大影响的dna甲基化事件,表明该基因是dna甲基化驱动基因。methylmix分析包括三个部分。首先,确定371个hcc样品的deg的甲基化数据与配对的基因表达数据之间的相关性,以鉴定导致基因表达发生变化的dna甲基化事件,仅选择通过相关性分析的基因进行进一步分析。其次,通过大量患者利用β混合模型来定义甲基化状态,从而排除了任意阈值的需要。第三,wilcoxon秩和检验用于比较371个hcc样品和50个相应的非肿瘤样品之间的dna甲基化状态。以0.05的q值为阈值。
步骤二:模型构建及模型验证
4)预测模型的构建和验证
在tcga的数据集中,单因素cox回归分析、最小绝对收缩和选择算子(lasso)、cox回归分析和多因素cox回归分析被用于评估dna甲基化驱动基因的表达与预后之间的关系,并鉴定与预后显著相关的dna甲基化驱动基因。通过使用来自多因素cox回归的系数作为权重,通过dna甲基化驱动基因的表达水平的线性组合,建立了基于dna甲基化驱动基因的风险评分预测模型。基于dna甲基化驱动基因的风险评分预测模型,以最佳风险评分为阈值,将hcc患者分为低风险患者和高风险患者。我们使用x-tile软件来找到最佳阈值。最佳阈值定义为在mantel-cox检验中产生最大χ2的风险评分。通过kaplan-meier生存曲线评估高风险患者和低风险患者之间的生存和复发差异,然后使用对数秩检验进行比较。我们使用geo数据库中的gse14520数据集来验证模型。通过kaplan-meier生存曲线评估高风险患者和低风险患者之间的生存和复发差异,然后使用对数秩检验进行比较。最后我们评估了该模型的诊断hcc的能力。
步骤三:实验验证
5)细胞培养
hcc细胞系hepg2购自atcc(
6)5-氮杂2'-脱氧胞苷(dac)处理
将培养的hepg2细胞用5μm/l5-氮杂2'-脱氧胞苷(dac)(sigma-aldrich,货号:a3656-5mg)处理120小时,并且由于dac不稳定性,每天更换培养基。对于涉及dac处理的实验,将二甲基亚砜(dmso)用作对照处理。收获细胞以提取基因组dna和总rna以分析dna甲基化和目的基因表达。
7)dna提取和dna甲基化分析
设计测序引物以包括cpg位点在转录起始位点0.5kb之内的片段。等于或低于15%的甲基化水平被认为与背景没有区别,而15%或更高的甲基化水平表示甲基化处理成功。我们使用
8)使用定量实时聚合酶链反应(qrt-pcr)验证mrna
根据制造商的说明,使用trizol试剂(thermofisher,货号:15596026)从培养的癌细胞中提取总rna。cdnareversetranscriptionkit(toyobo,货号:fsq-101)用于逆转录rna,而sybrgreenpcrkit(appliedbiosystems,货号:4368708)用于扩增所得的cdna。用quantstudio5real-timepcrsystem(appliedbiosystems;thermofisherscientific)检测样品。每个实验至少进行3次。采用2-δδct方法来计算相对于管家基因gapdh的基因表达。
- 还没有人留言评论。精彩留言会获得点赞!