一种治疗弱视的药物组合物的制作方法
本发明具体涉及一种治疗弱视的药物组合物。
背景技术:
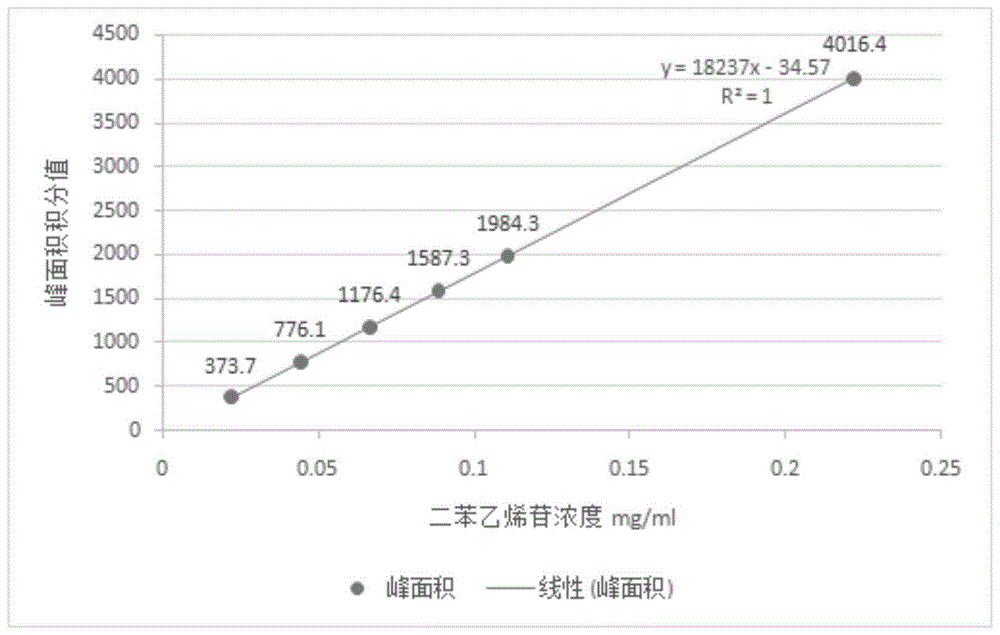
:弱视是眼部无器质性病变、矫正视力低于0.9,其为临床常见的儿童眼病,多始发于婴幼儿时期,由于各种原因如知觉、运动、传导及视中枢等原因未能接受适宜的视刺激,使视觉发育受到影响而发生的视觉功能减退的状态,主要表现为视力低下及双眼单视功能障碍。目前治疗弱视以西医为主,药物有左旋多巴和胞二磷胆碱应用于临床。多巴胺对成年人的副作用已很清楚,患者出现恶心呕吐、食欲减退、腹胀、心慌、头晕等不良反应,对小孩长期使用副作用更甚;胞二磷胆碱可能诱发癫痫发作,因此寻求中医药治疗儿童弱视成为一种更安全,更有效的方法。现已有关于治疗弱视的中药组合物问世,但多以外用辅助治疗为主,如专利cn108404052a公开了一种防治近视、弱视的药物组合物,其就是通过外敷达到治疗眼疾的目的,目前还缺乏口服的,效果显著的治疗弱视,尤其针对儿童弱视的中药组合物。技术实现要素:为解决上述问题,本发明提供了一种治疗弱视的药物组合物,它是由下述重量配比的原料药制备而成:何首乌117-217份、肉苁蓉117-217份、熟地黄117-217份、枸杞子117-217份、菟丝子117-217份、泽泻117-217份、丹参117-217份、川芎70-130份、陈皮70-130份。进一步地,它是由下述重量配比的原料药制备而成:何首乌167份、肉苁蓉167份、熟地黄167份、枸杞子167份、菟丝子167份、泽泻167份、丹参167份、川芎100份、陈皮100份。进一步地,所述何首乌为制何首乌;所述菟丝子为盐菟丝子。进一步地,它是由原料药的药粉、或原料药的水或有机溶剂提取物为活性成分,加上药学上可接受的辅料制备而成的制剂。更进一步地,所述制剂为口服制剂。更进一步地,所述口服制剂为颗粒剂、膏剂、散剂、丸剂或溶液剂,优选颗粒剂。本发明还提供了一种前述药物组合物的制备方法,它包括以下步骤:(1)按配比称取原料药;(2)加8~20倍量(w/v,g/ml)水回流提取,过滤,滤液浓缩成浸膏,加糊精混匀,干燥,粉碎,制粒,再干燥,过筛即得。进一步地,步骤2)所述加10倍量水回流提取3次,每次0.5小时;所述过滤为过200目滤网;所述浓缩为真空度-0.08~-0.1mpa,温度65℃减压浓缩;所述制粒是用80%酒精湿法制粒;所述干燥为60℃常压干燥;所述过筛是过一至五号筛。进一步地,所述浸膏60℃相对密度1.35~1.40;所述浸膏与糊精的质量比为1:1。本发明最后提供了一种前述药物组合物在制备治疗弱视的药物中的用途。进一步地,所述药物是治疗儿童弱视的药物。本发明药物组合物中,制何首乌为君药,滋肾水益肝血;熟地、枸杞子、肉苁蓉、盐菟丝子共为臣药,协助君药共奏补肝肾益精血之功,其中熟地黄能养肝补肾,精血并补,为滋养先天,治疗精血不足所致的目暗不明之要药;枸杞子具有滋补肝肾,养肝明目作用;配以补阳不燥,药力和缓之肉苁蓉以补肾阳益精血,于阴中求阳,以达到阴平阳秘;盐菟丝子能补肾益精,养肝明目益脾,既可补阳又可益阴,具有温而不燥,补而不滞的特点,为平补肝肾之良药。川芎、丹参为佐药,川芎为血中之气药,活血行气,通达气血;丹参功擅活血祛瘀,通行血脉,二药合用,使补中有动,动静兼顾,补而不滞,使肝肾之精血,循目中幽深之脉道,通达于目中,涵养目窍,共奏活血通络之功。泽泻、陈皮为使药,泽泻能泻肾,使补中有泻,此取其以泻为补之意,为防止滋补之品产生滞腻之弊;补益药物性质多粘腻,妨碍消化,故加少许陈皮,理气运脾,此二药在方中用量较小为使药。全方药物集中归经于肝肾,滋补肝肾兼通经络,补阳又可益阴,阴平阳秘,动静兼顾,使补而不滞,补中有泻,滋而不腻,协调阴阳。既考虑到眼部的生理特点,又兼顾了儿童稚阴稚阳之体。诸药合用,共奏培补肝肾,活血通络之功能,符合中医理法方药用药原则。经临床试验证明:本发明药物组合物具有治疗弱视的作用,尤其在治疗儿童弱视方面效果显著,具备实际应用推广价值。显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。附图说明图1二苯乙烯苷标准曲线图2二苯乙烯苷对照品色谱图图3样品溶液色谱图图4阴性溶液色谱图图5毛蕊花糖苷标准曲线图6毛蕊花糖苷对照品色谱图图7样品溶液色谱图图8阴性溶液色谱图图9临界相对湿度具体实施方式实施例1、本发明药物组合物的制备配方:制何首乌167g、肉苁蓉167g、熟地黄167g、枸杞子167g、盐菟丝子167g、泽泻167g、丹参167g、川芎100g、陈皮100g。制备方法:取上述九味饮片,加水回流提取煎煮三次(每次加10倍量水),每次0.5小时,200目滤网过滤,减压浓缩(温度65℃,真空度-0.08~-0.1mpa)成相对密度1.35~1.40(60℃)的浸膏,加入与浸膏等量的糊精,进行常压干燥(60℃),将干燥后的所得物进行打粉,粉碎,用80%酒精湿法制粒,60℃干燥,过筛,收集一至五号筛的颗粒,即得1000g颗粒,每克颗粒相当于原生药材1.37g。实施例2、本发明药物组合物的制备配方:制何首乌117g、肉苁蓉117g、熟地117g、枸杞子117g、盐菟丝子117g、泽泻117g、丹参117g、川芎70g、陈皮70g。制备方法:取上述九味饮片,加水回流提取煎煮三次(每次加10倍量水),每次0.5小时,200目滤网过滤,减压浓缩(温度65℃,真空度-0.08~-0.1mpa)成相对密度1.35~1.40(60℃)的浸膏,加入与浸膏等量的糊精,进行常压干燥(60℃),将干燥后的所得物进行打粉,粉碎,用80%酒精湿法制粒,60℃干燥,过筛,收集一至五号筛的颗粒,即得。实施例3、本发明药物组合物的制备配方:制何首乌217g、肉苁蓉217g、熟地217g、枸杞子217g、盐菟丝子217g、泽泻217g、丹参217g、川芎130g、陈皮130g。制备方法:取上述九味饮片,加水回流提取煎煮三次(每次加10倍量水),每次0.5小时,200目滤网过滤,减压浓缩(温度65℃,真空度-0.08~-0.1mpa)成相对密度1.35~1.40(60℃)的浸膏,加入与浸膏等量的糊精,进行常压干燥(60℃),将干燥后的所得物进行打粉,粉碎,用80%酒精湿法制粒,60℃干燥,过筛,收集一至五号筛的颗粒,即得。以下通过试验例来说明本发明的有益效果。试验例1本发明制备工艺筛选1、剂型的选择本发明配方量较大,出膏量多,做成胶囊剂或片剂等固体剂型的成本较高;汤剂会给患者造成使用和携带不便,且稳定性差,难于保存;而颗粒剂不仅保留了汤剂吸收快、起效迅速的特点,且服用、携带方便,同时稳定性提高,符合临床用药的需要,所以选定颗粒剂为本发明组合物的剂型。2药材前处理本发明所用药材饮片均为药典收载品种,符合《中华人民共和国药典》(2015年版一部)有关要求。制首乌为制首乌药材切制成的不规则的厚片或块的饮片。肉苁蓉为肉苁蓉药材切制而成的不规则厚片的饮片。熟地黄为地黄药材加工而成并切制成不规则厚片的饮片。丹参为丹参药材切制而成的类圆形或椭圆形厚片的饮片。泽泻为泽泻药材切制而成的圆形或椭圆形厚片的饮片。川芎为川芎药材切制而成的不规则厚片饮片。盐菟丝子为菟丝子药材经过盐炙法而成的药材。枸杞子为类纺锤形或椭圆形的药材。陈皮为陈皮药材切制而成的不规则条状或丝状的饮片。3原料提取工艺研究根据本发明配方九味药材所含有效成分的理化性质进行提取工艺研究,方中制首乌所含成分二苯乙烯苷,肉苁蓉所含成分松果菊苷和毛蕊花糖苷,熟地所含成分毛蕊花糖苷,丹参所含成分丹酚酸类、丹参酮类以及挥发油类,盐菟丝子所含成分黄酮类,多糖类,枸杞子所含成分多糖类,泽泻所含成分萜类、糖类,陈皮所含成分黄酮类、生物碱类、挥发油类,川芎所含成分生物碱类、苯酚酞类、有机酸类均能溶于水,故采用水提。4原料药水煎煮提取工艺研究4.1实验方法根据汤剂的制备方法、药材所含成分的性质和加工的实际情况,药材采用饮片形式以水煎煮提取数次,即可充分浸出有效成分。因此,对加水量、煎煮时间和次数三个因素各设三个水平安排试验,因素水平表见表1。表1水提因素水平表由于制首乌为本发明组合物的君药,二苯乙烯苷是其主要成分,也是本发明组合物主要指标成分;肉苁蓉和熟地为本发明组合物臣药,毛蕊花糖苷是二者的共有成分;故以二苯乙烯苷的含量、毛蕊花糖苷的含量以及干膏的收率为指标进行综合评价。按处方比例称取药材82g,按上述因素水平表的试验条件进行提取,过滤,滤液浓缩并定容至800ml(0.1025g生药/ml),备用。4.2各评价指标的含量测定方法4.2.1二苯乙烯苷的含量测定二苯乙烯苷的含量测量:采用高效液相色谱法色谱条件:agilent1260型高效液相色谱仪,色谱柱:zorbaxeclipsexdb-c18(4.6×150nm,5-mircon),检测波长320nm,柱温30℃,流动相:乙腈-水(15:85),进样量10μl,流速1.0ml·min-1。对照品溶液的制备:精密称取2,3,5,4’-四羟基二苯乙烯-2-o-β-d-葡萄糖苷对照品(中国食品药品检定研究院,批号:110844-201713)11.1mg,置20ml容量瓶中,加稀乙醇溶解并稀释,得1.11mg/ml对照品溶液。供试品溶液的制备:精密吸取水提浓缩液4ml,置水浴锅上蒸干,精密加入稀乙醇25ml,使溶解,转移至圆底烧瓶中,称定重量,加热回流30分钟,放冷,再称定重量,用稀乙醇补足减失的重量,摇匀,静置,上清液滤过,取续滤液,过微孔滤膜(0.45μm),即得。阴性样品溶液的制备:为考察二苯乙烯苷含量测定有无干扰,需制备阴性样品。取缺制首乌的药材,按相同试验条件提取,精密量取提取液体积,按供试品溶液的制备项下方法同法制成阴性样品溶液。标准曲线及线性范围考察:照高效液相色谱法《中国药典》一部附录vid试验,精密吸取对照品溶液0.2,0.4,0.6,0.8,1,2ml,分别置于10ml量瓶中,加稀乙醇稀释并定容至刻度,得6个质量浓度的2,3,5,4’-四羟基二苯乙烯-2-o-β-d-葡萄糖苷对照品溶液,按色谱条件测定,测定结果见表2,以峰面积为纵坐标,质量浓度为横坐标(图1),并计算回归方程。表2二苯乙烯苷线性关系的考查c(mg/ml)进样量(μl)峰面积0.022210373.70.044410776.10.0666101176.40.0888101587.30.111101984.30.222104016.4结果得回归方程y=18237x-34.57(r=0.9999),表明2,3,5,4’-四羟基二苯乙烯-2-o-β-d-葡萄糖苷在0.0222~0.222mg·l-1范围内呈良好线性关系。二苯乙烯苷峰面积的自然对数与进样量的自然对数线性关系良好,又因该标准曲线不过原点,斜率小于100倍截距,故需采用外标两点法测定二苯乙烯苷含量。阴性干扰的判断:将阴性样品溶液、样品溶液、对照品溶液分别注入高效液相色谱仪,记录色谱图,见图2、图3、图4。从以上图谱可以看出,阴性样品在二苯乙烯苷峰处无干扰,因此以二苯乙烯苷为指标性成分进行考察。对照品稳定性考察:分别吸取对照品溶液10μl,于0、2、4、6、8、24h时按前述色谱条件进行测定,结果见表3,由表3可知对照品在24h内稳定性良好。表3二苯乙烯苷稳定性实验供试品稳定性考察:分别吸取供试品溶液(批号:170801)10μl,于0、2、4、6、8、24h时按前述色谱条件进行测定,结果见表4,从表4可知供试品在24h内稳定性良好。表4制首乌供试品稳定性实验二苯乙烯苷的转移率(%)=测得二苯乙烯苷的量(g)/制首乌原药材中二苯乙烯苷的量(g)*100%4.2.2毛蕊花糖苷的含量测定毛蕊花糖苷的含量测定:采用高效液相色谱法。色谱条件:agilent1260型高效液相色谱仪,色谱柱:diamonsilplus5μmc18-a(250×4.6mm))检测波长330nm,柱温30℃,以甲醇为流动相a,0.1%甲酸水溶液为流动相b,(梯度洗脱程序如下:0~20min,29.5a;20~21min,29.5→27a;21~35min,27a;35~37min,27→90a;37~43min,90a。),进样量10μl,流速1.0ml·min-1。对照品溶液的制备:精密称取毛蕊花糖苷对照品7.87mg(中国食品药品检定研究院,批号:111530-201713),置10ml容量瓶中,加50%甲醇溶解并稀释,得0.787mg/ml对照品溶液。供试品溶液的制备:精密吸取水提浓缩液20ml,置水浴锅上蒸干,精密加入50%甲醇50ml,使溶解,转移至100ml棕色容量瓶中,称定重量,浸泡30分钟,超声处理40分钟(功率250w,频率35khz),放冷,再称定重量,加50%甲醇补足减失的重量,摇匀,静置,取上清液,滤过,取续滤液,过微孔滤膜(0.45μm),即得。阴性样品溶液的制备:为考察毛蕊花糖苷含量测定有无干扰,需制备阴性样品。取缺肉苁蓉和熟地的药材,按试验提取,精密量取提取液体积,按供试品溶液的制备项下方法同法制成阴性样品溶液。标准曲线及线性范围考察:照高效液相色谱法《中国药典》一部附录vid试验,精密吸取对照品溶液0.1,0.2,0.3,0.4,0.5,1ml,分别置于10ml量瓶中,加50%甲醇稀释并定容至刻度,得6个质量浓度的毛蕊花糖苷对照品溶液,按色谱条件测定,测定结果见表5。以峰面积为纵坐标,质量浓度为横坐标(见图5),计算回归方程。表5毛蕊花糖苷线性关系的考查c(mg/ml)进样量(μl)峰面积0.007871090.80.0157410192.50.0236110281.60.0314810373.80.0393510471.20.078710990.5结果得得回归方程y=12686x-15.928(r=0.9992),表明毛蕊花糖苷在0.00787~0.0787mg·l-1范围内呈良好线性关系。毛蕊花糖苷峰面积的自然对数与进样量的自然对数线性关系良好,又因该标准曲线不过原点,斜率小于100倍截距,故需采用外标两点法测定毛蕊花糖苷含量。阴性干扰的判断:将阴性样品溶液、样品溶液、对照品溶液分别注入高效液相色谱仪,记录色谱图,见图6、图7、图8。从以上图谱可以看出,阴性样品在毛蕊花糖苷峰处无干扰,因此以毛蕊花糖苷为指标性成分进行考察。对照品稳定性考察:分析吸取对照品溶液10μl,于0、2、4、6、8、24h时按前述色谱条件进行测定,结果见表6,表6表明对照品在24h内稳定性良好。表6毛蕊花糖苷稳定性实验供试品稳定性考察:分析吸取供试品溶液(批号:170801)10μl,于0、2、4、6、8、24h时按前述色谱条件进行测定,结果见表7,表7表明供试品在24h内稳定性良好。表7肉苁蓉和熟地供试品稳定性实验毛蕊花糖苷的转移率(%)=测得毛蕊花糖苷的量(g)/肉苁蓉和熟地原药材中毛蕊花糖苷的量(g)*100%4.2.3.干膏的收率精密量取因素水平表下提取液一定量,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105℃干燥6小时,移至干燥器中,冷却30分钟,迅速精密称定重量,计算浸膏得率。4.3试验结果及分析根据熵权法将二苯乙烯苷的含量、毛蕊花糖苷的含量以及干膏的收率的权重系数分别定为0.3868、0.4345、0.1787,根据各权重系数计算综合得分,结果见表8。表8试验结果由直观分析结果可知,a3b1c3为最佳提取工艺,即加10倍量水,每次提取0.5h,提取3次。实验验证为了保证工艺的稳定性,根据所筛选的最佳工艺条件进行实验验证,结果见表9。表9提取工艺验证试验验证试验结果表明,所筛选的提取工艺条件稳定可行。4.4纯化工艺的研究4.4.1水提取液除杂通过计算二苯乙烯苷与毛蕊花糖苷的保有率以及干膏率,从而对过滤与离心两种方法进行考察。实验方法:按处方称取药材,依所筛选的工艺条件进行提取,并量取3份不同体积的溶液(浓缩至640ml,240ml,200ml,200ml)的分别进行不处理(既不过滤也不离心)、过滤、离心(4000r/min,10min)分别测定二苯乙烯苷、毛蕊花糖苷和干膏收率,结果见表10。表10不同除杂方式效果比较以上结果表明:使用过滤的方式进行纯化时,二苯乙烯苷和毛蕊花糖苷的保有率均比离心的方式效果好,而出膏率几乎无变化,因此选用过滤的方式进行除杂。4.4.2水提取液的浓缩工艺水提取液经过滤后,分别采用不处理(既不常压浓缩也不减压浓缩)、常压浓缩、减压浓缩3种处理方法进行处理样液。3次水提取液共8770ml,其中770ml不进行处理;4000ml进行常压浓缩,浓缩至250ml;4000ml进行减压浓缩,减压浓缩温度控制在65℃、真空度-0.08~-0.1mpa,浓缩至310ml。测定浓缩前后二苯乙烯苷和毛蕊花糖苷的保有率变化,结果见表11。表11水提取液浓缩前、减压浓缩、常压浓缩含量的变化结果表明,采用减压浓缩,有效成分损失小。4.4.3水提取液的干燥工艺水提取液经过过滤,减压浓缩至浸膏相对密度1.35~1.40(温度控制在60℃),在60℃下的常压干燥环境下进行干燥,结果见表12。表12干燥后含量的变化干燥二苯乙烯苷的转移率(%)毛蕊花糖苷的转移率(%)常压干燥(60℃)69.7343.984.5成型工艺研究4.5.1制粒①辅料筛选采用湿法制粒进行制粒、干燥。取浸膏,加入糊精混匀,干燥,粉碎,进行湿法制粒,酒精的浓度为80%,在60℃温度下进行干燥,过筛,收集一至五号筛的颗粒,测定粒度,并观察外观,结果见表13。表13辅料筛选从表13的结果进一步确定湿法制粒的条件是:取浸膏,加入等量糊精,干燥,粉碎,用80%浓度的酒精进行湿法制粒,干燥温度为60℃,过筛,收集一至五号筛的颗粒。②颗粒吸湿性考查分别取一步制粒所得颗粒7份,每份约2g,置于已干燥至恒重的称量瓶中,精密称定,置于7种不同相对湿度(30~90%)的干燥器中(称量瓶盖揭开),在25℃的培养箱中放置7天,取出称量瓶,加盖后精密称定,计算含水量,以水分百分含量为纵坐标,相对湿度为横坐标作曲线。结果见表14、图9。表14各种相对湿度条件下颗粒吸湿结果结果表明,颗粒的临界相对湿度为68%,饱和吸湿率为42.36%,因此,要求生产净化间(包括制粒车间和分装车间)的相对湿度应控制在68%以下,颗粒的包装和贮存也应防潮。③颗粒流动性考察为保证工业化生产顺利进行,分装时剂量准确,要求颗粒的流动性较好,采用测定休止角的方法考察颗粒流动性,结果见表15。表15颗粒休止角测定结果结果表明:该颗粒休止角小于40°,流动性良好,易于分装。4.5.2包装材料的选择本发明颗粒具有一定的吸湿性,实验用中试样品三批(17001、17002、17003)以铝塑复合袋包装,于室温(10~40℃)、相对湿度50~90%考察3个月。结果颗粒的水分符合要求,外观干燥整洁,表明铝塑复合袋包装能够满足贮存要求。4.6分装颗粒分装机分装,分装规格为20g/袋。5中试研究5.1中试情况按上述确定的工艺路线及条件中试,生产三批样品,主要生产数据见表16。表16中试生产数据5.2中试产品质量检查按照《中国药典》颗粒剂通则及自拟药品质量标准对中试样品三批(17001、17002、17003)进行质量检查,结果见表17。表17三批中试样品质量检查中试结果表明:本发明连续生产的稳定性好,工艺条件合理可行,符合大生产的要求。试验例2临床试验1试验设计1.1样本量确定由于儿童弱视的相关研究结论不易统一,因而尚缺乏计算本发明样本的依据。试验时考虑到研究周期和经费的限制,采用小样本研究,每组30例,考虑脱落等因素扩大10%样本量,即总样本量66例,每组33例。1.2随机方法符合儿童弱视诊断标准,并按纳入、排除标准确定的病例,随机分为治疗组和对照组,两组按1∶1设立,样本量设计各33例,采用多中心随机对照及盲法试验。四川省中医药科学院中医研究所收集病例16例,四川省中西医结合医院收集病例20例,成都市第二人民医院收集病例30例。采用完全随机方案,聘请专人以随机数字表法设计。设计完成后装入按序列编码的、密封的、不透光的信封中,交研究负责人控制,当筛选到合格病例后,由负责人进行随机分组。1.3盲法设计根据中医临床研究的特点,实行治疗操作、观测记录、统计分析三分离。为了贯彻盲法精神,拟采取以下措施:①避免向患者透露分组、治疗等的相关详细信息(涉及患者知情的内容除外)。②随机分组方案由临床研究人员以外的专人负责。③由治疗操作以外的专业技术人员1~2名进行相关资料的收集、评判、整理、记录等工作。④聘请专业统计分析人员进行统计学处理,病例分组情况对其保密。1.4对照方法的选择设立实验对照,即以常规基础治疗为对照。2研究对象(病例选择)2.1诊断标准2.1.1西医诊断标准参照1996年4月中华眼科学会全国儿童弱视斜视防治学组工作会议制定的标准,弱视的定义是:凡眼部无明显器质性病变,以功能性因素为主引起的远视力≤0.8且不能矫正者,称为弱视。按程度分为:①轻度弱视:矫正视力0.6~0.8。②中度弱视:矫正视力0.2~0.5。③重度弱视:矫正视力≤0.1。按类型分为:①斜视性弱视:患者有斜视或曾有过斜视。②屈光参差性弱视:患者双眼屈光度相差球镜≥1.5d,柱镜≥1.0d。③屈光不正性弱视:为双侧性,发生于未戴过矫正眼镜的高度屈光不正患者,双眼视力相等或接近,远视≥3.00d,近视≥6.00d,散光≥2.00d。④形觉剥夺性弱视:在婴幼儿期由于屈光间质混浊、上睑下垂遮盖瞳孔、不适当地遮盖等引起的视功能障碍。⑤其他。包括新生儿视网膜或视路出血引起的器质性弱视,眼球震颤性弱视和色盲性弱视。2.1.2中医辩证标准因目前无统一儿童弱视中医辩证标准,参照1994年国家中医药管理局发布的中华人民共和国中医药行业标准《中医病症诊断疗效标准》中“能近怯远”肝肾亏虚证:远视力下降,或伴有眼前黑花飞舞。舌淡红,少苔,脉无特殊变化。2.2纳入标准①符合西医弱视诊断分类标准的前三类(即斜视性弱视,屈光参差性弱视,屈光不正性弱视);②符合西医诊断标准(即轻到中度弱视);③符合中医辨证标准(即肝肾亏虚、经脉不通证型的病例);④年龄3~12周岁;⑤志愿受试并签署知情同意书。2.3排除标准①合并有心血管、脑血管、肝、肾和造血系统等严重全身性疾病及精神病患者;②长期服用其它有关治疗药物或使用其它治疗方法,未能终止者;③患眼合并其它眼病,影响疗效判定者;④未按规定用药,无法判定疗效,或资料不全等影响疗效或安全性判断者。2.4剔除、脱落标准2.4.1剔除标准①纳入后发现不符合纳入标准者;②纳入后未接受过试验方案所规定的治疗措施者。2.4.2脱落标准①未完成试验而中途退出者;②未按规定方案治疗,或合并使用其他疗法或药物而无法判定疗效者;③出现不良事件或不良反应者。2.5中止标准①严重不良事件或不良反应者;②治疗无效者;③设计方案或试验中出现重大问题,无法判定疗效者。3治疗方案(病例处置)3.1干预措施3.1.1治疗组:采用基础治疗+内服中药(1)常规基础治疗方案如下:①验光配镜:使用1%阿托品眼膏一日三次,共三日,麻痹睫状肌后验光,确定戴镜度数,配戴合适的眼镜矫正屈光不正。配镜原则:斜视性弱视:内斜视者,远视全部矫正,近视给予能看1.0时的所需的最低度数;外斜视者,近视全部矫正,远视低度者(小于+3.00球)可不给,大于+3.00球者,可减去+1.00~+2.00球;斜视合并散光的患者,不论散光的正与负都应全部矫正。屈光不正性弱视:给予全部矫正。屈光参差性弱视者,如能接受应全部矫正,如不能接受,应适当缩小参差。②遮盖主眼:根据弱视情况采取单眼遮盖或交替遮盖。4岁以下,遮盖主眼4天,然后再遮盖弱视眼1天;4岁以上,遮盖主眼6天,然后再遮盖弱视眼1天,以避免发生遮盖性弱视的危险。③精细用眼工作:遮盖主眼或弱视眼,强迫使用弱视眼,穿珠子,穿针,刻剪纸,描画等,以消除主眼对弱视眼的抑制。每天一次,每次30分钟。疗程:4周为一疗程,共治疗2疗程。(2)内服中药本发明药物组合物,其组成:制何首乌167g、熟地黄167g、枸杞子167g、肉苁蓉167g、盐菟丝子167g、川芎100g、丹参167g、泽泻167g、陈皮100g。(中药配方颗粒剂,四川新绿色药业科技发展股份有限公司,生产许可证号:川z20090440,按实施例1方法制备),6到12岁患儿一日一剂,分三次服,每次100ml,6岁以下患儿两日一剂,分三次服,每次50ml。疗程:4周为一疗程,共治疗2疗程,每周服药4天,停药休息3天(若治疗期间出现感冒发热、腹泻等情况,暂停治疗,恢复后再进入治疗,疗程接着计算)。3.1.2对照组:采用基础治疗方法、疗程同治疗组。3.2治疗分配在三个中心,采用完全随机方案,聘请专人以随机数字表法设计。方案设计完成后装入按序列编码的、密封的、不透光的信封中,交研究负责人控制,当筛选到合格病例后,由负责人进行随机分组。随机分为治疗组和对照组,两组病例按1∶1设立,样本量设计各33例,采用多中心随机对照及盲法试验。3.3合并用药①除规定用药处,试验期间禁止使用影响观察试验药物疗效的中药和西药。②合并疾病所必须继续服用的药物,或其他治疗必须在病例报告表中记录药名(或其它疗法名)、用量、使用次数和时间等,以便总结时加以分析和报告。4质量控制与保证措施为保证本发明药物的临床试验的顺利进行,同时保证研究的客观性、真实性和可靠性,拟制定质控与质保措施如下:4.1组织措施①拟设质控专员1名,对整个研究过程中的各个环节进行质量控制和监督,检查、确认所有研究数据的记录与报告以及病例报告表填写的正确与完整,并保证与原始资料一致。②设试验负责人1名;治疗操作人员各固定2~3名,观察记录人员各固定1~2名;聘请随机分组方案设计人员1名,统计分析人员1名。所有研究人员实行岗位责任、分工合作制。4.2技术措施①人力培训:研究工作正式开展之前,对所有研究人员进行集中统一培训,熟悉并掌握本临床实施方案和要求等。②严格、一致地执行研究方案:在研究工作的各个环节均应严格、一致地执行研究方案,避免参入主观意志,随心所欲。③研究对象的管理:签署知情同意书,保证治疗依从性,向受试者强调观察期内禁止采用其他疗法或服用相关药物;对自愿脱落者,应通过电话、信件或上门随访,询问原因,调查其后的经过,并进行受试者中途撤除研究时的临床评价。5资料整理、数据管理及统计分析5.1资料整理试验结束后,由资料记录人员将病例报告表上的原始观察结果转录在统一印制并编有页码的实验记录本上。涉及不符合规范的病例资料,如脱落、中止及无法判断疗效者,按下述原则处理:符合剔除标准而剔除者,不记入统计;对未完成试验而中途退出者,若因无效而退出者,记入疗效(判无效)统计;对出现不良事件或不良反应者,不记入疗效统计,而记入不良反应统计;对设计方案或试验中出现重大问题而中止者,不记入统计。5.2数据管理由数据管理人员逐份审核病例报告表(crf),进行完整性和逻辑性检查,对缺损数据及有逻辑错误的数据逐项登记,并向记录者发出调查问卷予以完善、更正。然后采用spss11.5软件编制数据录入程序进行数据录入与管理,采用双人校对录入,一人读取数据,另一人输入,读取者再予监督,对不一致的结果值,逐项核对原始crf予以更正。数据录入结束后,由监察员随机抽取10%的crf与数据库进行人工核对,确保数据一致,确定有无缺失值或异常值,修改错误数据。5.3统计分析整个数据库数据修改、确认后,包含按计划完成且符合研究方案要求的病例数据集,对其进行锁定、封盲。然后交由专业人员独立进行统计分析,分析结束后予以揭盲。统计分析:用spss17.0软件分析计算。①描述性分析:计量资料和半计量资料用(症状评分)用x±s表示;计数资料用率或构成(比)表示。②假设检验:计量资料符合正态分布用t检验(方差不齐时用校正t’检验),不符合正态分布用mann-whitneyu检验(m-w检验);半计量资料用非参数相关或独立样本的m-w检验;非等级计数资料用χ2检验;等级计数资料用非参数相关或独立样本的m-w检验;“不良反应”结果的分析,用logistic回归分析。显著性检验水准α=0.05,p值取单侧值。6伦理学原则本发明临床研究已通过四川省第二中医医院伦理委员会的审查,同意开始临床研究。7总结与资料保存7.1总结建立临床资料数据库,统一进行数据处理,由统计分析人员进行统计和综合全部资料进行统计。完成“临床试验总结报告”。7.2资料保存试验方案、知情同意书(样式)、原始门诊病历、病例报名表等。8总体性能指标8.1指标选择8.1.1效应指标①视力检测:患者每次就诊时,用国际标准视力表检测双眼视力,记录结果。②注视性质:患者每次就诊时,用直接检眼镜检测注视性质,记录结果。③起效时间:以视力提高2行的时间为起效时间。8.1.2不良反应指标包括恶心呕吐、食欲减退、腹胀、心慌、头晕、体位性低血压等〔13〕。以上指标的分级评定方法:轻度:略有不适,可耐受,不必处理1分中度:有不适,较难耐受,需对应处理2分重度:严重不适,难耐受,应停止试验并进行及时处理或抢救3分8.1.3安全性指标对正常生理状态干扰指标,包括血常规、肝肾功检查。8.1.4观测时点本临床试验设立治疗前、治疗第一个疗程末、治疗第二个疗程末,治疗结束后半年末,共4个时点。若观察期未到二个疗程因临床基本痊愈而停止治疗,则观察纪录至实际结束时点。随机对照观察期结束后,停止各组相应治疗,进入洗脱期,消除各治疗措施对受试者的影响。在停止治疗后半年、一年(洗脱期后点)对受试者进行随访。随访的指标包括视力检测和注视性质,评定方法同观测期。8.2临床评价8.2.1疗效评价(1)临床疗效判定疗效评价标准:根据1996年4月中华眼科学会全国儿童弱视斜视防治学组工作会议通过的弱视治疗疗效评价标准。①痊愈:经过3年随访,视力仍保持正常(评定痊愈的话必须随访3年以上,因本研究周期为两年,此项指标暂时不纳入本次研究的疗效评价标准);②基本痊愈:矫正视力提高至0.9或以上;③进步:视力提高2行或2行以上;④无效:视力退步、不变或提高仅1行。(2)注视性质的分析、评价。(3)起效时间的分析、评价。8.2.2不良反应评价(1)不良反应因果判定分肯定有关、可能有关、无法确定、可能无关和肯定无关5级。其判定依据下列5项:a.开始治疗时间与可疑不良反应出现有无合理的先后关系。b.可疑不良反应是否符合该疗法已知的不良反应类型。c.可疑不良反应能否可用合并疗法、病人临床状况或其他药物的影响来解释。d.停止治疗后可疑不良反应是否消失或减轻。e.再次施加该疗法后,是否重新出现同样的反应。判断标准:按以上5项顺序判断,见下表18。表18不良反应判断标准注:+:肯定;-:否定;±:难以肯定或否定;?:情况不清(2)不良反应的发生率不良反应的发生率=(a+b+c)的病例数÷全部观察病例数×100%8.2.3安全性评价1级:安全,无任何不良反应,安全性指标检查无异常。2级:比较安全,有较度不良反应,不需做任何处理可继续给药,安全性指标检查无异常。3级:有安全性问题,有中等程度的不良反应,或安全性指标检查有轻度异常,做处理后可继续给药。4级:因严重不良反应中止试验,或安全性指标检查明显异常。9研究结果9.1基线情况研究对象为2014年7月至2016年2月在四川省中医药科学院中医研究所眼科、四川省中西医结合医院五官科、成都市第二人民医院眼科门诊诊断为儿童弱视患儿。三个中心研究共纳入76例,采用随机数字表法将纳入的受试者随机分为2组,受试过程中脱落2例,共74例患儿(134只眼)完成治疗。治疗组(38例69只眼),男性19例,女性19例;3-6岁18例34只眼,7-12岁20例35只眼,平均年龄(6.25±2.63)岁;屈光不正性弱视34例65只眼,屈光参差性弱视4例4只眼;轻度弱视37只眼,中度弱视32只眼;平均矫正视力(0.55±0.13);平均病程(12.93±16.84)月。对照组(36例65只眼),男性16例,女性20例;3-6岁19例36只眼,7-12岁17例29只眼,平均年龄(5.65±2.53)岁;屈光不正性弱视31例60只眼,屈光参差性弱视5例5只眼;轻度弱视35只眼,中度弱视30只眼;平均矫正视力(0.58±0.10);平均病程(9.56±16.68)月。两组病例的性别、年龄、病程、治疗前矫正视力分别经χ2检验、非参数mann-whitneyu检验,均p>0.05,组间具有可比性。详见表19~表22。本临床试验已通过四川省中医药科学院中医研究所伦理委员会批准,所有受试者均进行知情同意并签署知情同意书。表19两组性别分布对比经χ2检验,p>0.05。表20两组年龄分布对比经mann-whitneyu检验,p>0.05。表21两组平均病程分布对比经mann-whitneyu检验,p>0.05。表22治疗前各指标对比经mann-whitneyu检验,治疗前各项指标对比均p>0.05。9.2两组患儿治疗前后视力比较表23所示,两组患儿治疗后视力均明显提高,经mann-whitneyu检验,治疗组自身治疗前后视力对比,z=-6.950,p<0.05;对照组自身治疗前后视力对比,z=-5.988,p<0.05。治疗后治疗组明显优于对照组(z=-3.326,p<0.05)。两组间视力变化值对比,z=-4.892,p<0.05。表23两组弱视患儿治疗前后视力比较注:与本组治疗前比较,△p<0.05;与对照组治疗后比较,*p<0.059.3两组患儿临床疗效比较表24所示,治疗组总有效率85.5%,对照组总有效率50.8%。经mann-whitneyu检验,z=-3.790,p<0.05,两组在疗效上有统计学差异,且治疗组优于对照组。表24两组弱视患儿临床疗效比较﹝眼(%)﹞9.4不同年龄段两组患儿临床疗效比较表25所示,3-6岁患儿,治疗组有效率79.4%,对照组有效率25.0%。经mann-whitneyu检验,z=-4.280,p<0.05,即3-6岁患儿,两组在疗效上有统计学差异,且治疗组优于对照组。7-12岁患儿,治疗组有效率91.4%,对照组有效率79.3%。经mann-whitneyu检验,z=-1.363,p>0.05,即7-12岁患儿,两组在疗效上无统计学差异。表25不同年龄段两组弱视患儿临床疗效比较﹝眼(%)﹞9.5不同程度弱视两组患儿临床疗效比较表26所示,轻度弱视,治疗组有效率86.5%,对照组有效率74.3%。经mann-whitneyu检验,z=-1.355,p>0.05,即两组在疗效上无统计学差异;中度弱视,治疗组有效率84.4%,对照组有效率30.0%。经mann-whitneyu检验,z=-4.380,p<0.05,两组在疗效上有统计学差异,且治疗组优于对照组。表26不同弱视程度两组弱视患儿临床疗效比较﹝眼(%)﹞9.6两组患儿治疗后视力进步的起效时间比较表27所示,治疗组视力进步的平均起效时间2.029±1.42月,对照组3.77±1.46月。经mann-whitneyu检验,z=-6.036,p<0.05,两组治疗后视力进步的起效时间有统计学差异,且治疗组短于对照组。表27两组视力进步的起效时间比较﹝眼(只)﹞9.7不同年龄段两组患儿治疗后视力进步的起效时间比较表28所示,3-6岁患儿,治疗组视力进步的平均起效时间1.20±0.41月,对照组2.50±1.69月。经mann-whitneyu检验z=-3.425,p<0.05,两组治疗后视力进步的起效时间有统计学差异,且治疗组短于对照组;7-12岁患儿,治疗组治疗后视力进步的平均起效时间1.29±0.54月,对照组2.56±1.59月。经mann-whitneyu检验,z=-2.948,p<0.05,两组治疗后视力进步的起效时间有统计学差异,且治疗组短于对照组。表28不同年龄段两组视力进步的起效时间比较9.8不同程度弱视两组患儿治疗后视力进步的起效时间比较表29所示,轻度弱视,治疗组视力进步的平均起效时间2.189±1.41月,对照组3.305±1.39。经mann-whitneyu检验,z=-3.359,p<0.05,两组治疗后视力进步的起效时间有统计学差异,且治疗组短于对照组;中度弱视,治疗组视力进步的平均起效时间1.843±1.43月,对照组4.296±1.38。经mann-whitneyu检验,z=-4.792,p<0.05,两组治疗后视力进步的起效时间有统计学差异,且治疗组短于对照组。表29不同弱视程度两组视力进步的起效时间比较﹝眼(只)﹞9.9两组患儿不良反应情况三个中心,两组患儿均未出现明显不良事件,所有完成的检查中,指标均在正常范围内。提示本研究采用培补肝肾活血通络法治疗儿童弱视具有较好的安全性。10结论(1)治疗儿童弱视,培补肝肾活血通络法和单纯基础治疗,两种方法均有效。结果显示治疗组临床疗效(总有效率85.5%),明显高于对照组(总有效率50.8%),(p<0.05),提示在总体疗效上,采用培补肝肾活血通络法治疗儿童弱视优于单纯基础治疗。(2)3-6岁儿童弱视,治疗组临床疗效(有效率79.4%),明显高于对照组(有效率25.0%),(p<0.05),提示治疗3-6岁儿童弱视,采用培补肝肾活血通络法优于单纯基础治疗。(3)7-12岁儿童弱视,培补肝肾活血通络法(有效率91.4%)与单纯基础治疗(有效率79.3%),临床疗效无显著性差异(p>0.05)。(4)轻度儿童弱视,培补肝肾活血通络法(有效率86.5%)与单纯基础治疗(有效率74.3%),临床疗效无显著性差异(p>0.05)。(5)中度儿童弱视,治疗组临床疗效(有效率84.4%),明显高于对照组(有效率30.0%),(p<0.05)。提示治疗中度儿童弱视,采用培补肝肾活血通络法优于单纯基础治疗。(6)视力进步的平均起效时间,治疗组(2.029±1.42月),明显短于对照组(3.77±1.46月),(p<0.05)。提示采用培补肝肾活血通络法较单纯基础治疗,总体上视力进步起效更快。(7)3-6岁儿童弱视,治疗组视力进步的平均起效时间(1.20±0.41月),明显短于对照组(2.50±1.69月),(p<0.05);7-12岁儿童弱视治疗组(1.29±0.54月),明显短于对照组(2.56±1.59月),(p<0.05)。提示不同年龄段儿童弱视,采用培补肝肾活血通络法较单纯基础治疗,均起效更快。(8)轻度儿童弱视,治疗组视力进步的平均起效时间(2.189±1.41月),明显短于对照组(3.305±1.39月),(p<0.05)。中度儿童弱视,治疗组(1.843±1.43月),明显短于对照组(4.296±1.38月),(p<0.05)。提示不同程度儿童弱视,采用培补肝肾活血通络法较单纯基础治疗,均起效更快。(9)两组治疗过程中均未出现明显不良反应。(10)培补肝肾活血通络法治疗儿童弱视,发挥了中医中药的优势,创新中医在此病理论上的认识,在治法上切实有效、安全可靠、无不良反应,临床疗效优于单纯基础治疗,无痛苦、易于被患儿及家长接受,无需要其他设备的投入,起效更快,能缩短疾病疗程,有临床应用价值。综上,本发明药物组合物,既考虑到眼部的生理特点,又兼顾了儿童稚阴稚阳之体,组合物中各药味合用,共奏培补肝肾,活血通络之功能,符合中医理法方药用药原则,经临床试验证明:本发明药物组合物具有治疗弱视的作用,尤其在治疗儿童弱视方面效果显著,具备实际应用推广价值。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1