铜表面掺杂VIII族金属促进甲烷分解的模拟仿真方法与流程
本发明属于催化甲烷分解技术领域,具体涉及一种铜表面掺杂viii族金属促进甲烷分解的模拟仿真方法。
背景技术:
甲烷是天然气的主要成分,当今世界天然气的产量持续增加,使得甲烷转化为高附加值的化学品成为催化领域的热门课题。例如,甲烷重整制合成气、甲烷氧化制甲醇、甲烷裂解制氢气等。另一方面,甲烷也是温室气体之一,会导致地球表面温室效应不断增加,甲烷的催化转化有利于缓解温室效应。然而,甲烷是一种非常稳定的分子,打破甲烷中的c-h键需要很高的热量,因此,甲烷的活化具有很大挑战性。
在常用的铜基催化剂表面掺杂过渡金属,通过两种金属的协同作用可以降低反应的活化能,使得甲烷的活化在更低的温度下发生,有效的节约能源,筛选出一种合适的金属进行掺杂具有重要意义。现有技术中大多采用实验的方法制备研究催化剂,当需要研究多种过渡金属的掺杂时,需要花费很多的时间、人力和物力。
密度泛函理论经过多年的发展和改进,形式更加精确,计算程序和算法更加高效,已经发展为模拟计算领域的一种重要工具和手段,其计算精度能够满足多种研究工作的需求,在材料、物理、化学等领域中有广泛的应用。用该方法来模拟甲烷分解过程,筛选一种合适的能有效促进甲烷分解的催化剂对实验研究具有很重要的指导意义。
技术实现要素:
本发明的目的在于解决现有技术中甲烷分解反应所需能量较高,需筛选一种合适的金属进行掺杂以降低反应活化能,但是实验方法耗时耗力的问题,提供一种铜表面viii族金属促进甲烷分解的模拟仿真方法。
本发明所涉及的一种铜表面掺杂viii族金属促进甲烷分解的模拟仿真方法,其特殊之处在于,包括以下步骤:
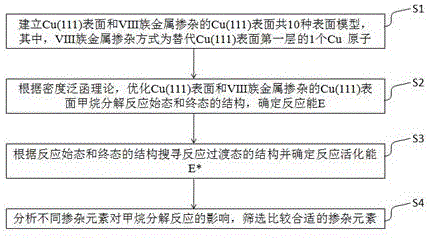
s1:建立cu(111)表面和viii族金属掺杂的cu(111)表面共10种表面模型,10种表面模型分别以fe-cu、co-cu、ni-cu、ru-cu、rh-cu、pd-cu、pt-cu、os-cu、ir-cu、cu表示,其中,viii族金属掺杂方式为替代cu(111)表面第一层的1个cu原子;
s2:根据密度泛函理论,优化cu(111)表面和viii族金属掺杂的cu(111)表面甲烷分解反应始态和终态的结构,确定反应能e。反应能e的计算方法为,用基元反应终态的能量减去始态的能量。如果数值为正值,表示该基元反应为吸热反应,如果数值为负值,表示该基元反应为放热反应;
s3:根据反应始态和终态的结构搜寻反应过渡态的结构并确定反应活化能e*,活化能的计算方法为,用过渡态的能量减去反应始态的能量,数值越大表示反应越难发生;
s4:分析不同掺杂元素对甲烷分解反应的影响,筛选比较合适的掺杂元素。
进一步地,所述步骤s1的cu(111)表面模型构建方法为,选取p(3×3)超晶胞构建模型,选取4层原子厚度,选取真空层厚度为15å,把第一层的一个cu原子替换为viii族金属即可得到viii族金属掺杂的cu(111)表面模型。
进一步地,所述步骤s2中甲烷分解反应包含ch4→ch3+h,ch3→ch2+h,ch2→ch+h,ch→c+h共4个基元反应,对每个基元反应的始态和终态在10种表面模型上的结构进行优化,优化过程中参数设置为,电子与电子之间的交换关联能均采用gga泛函数进行校正,平面波截断能取400ev,布里渊区积分k点取值3×3×1,电子自洽迭代和离子弛豫的收敛标准分别设置为能量收敛至10-5ev和原子力收敛至0.02ev/å。
进一步地,在结构优化过程中,催化剂模型下部两层原子固定,顶部两层原子及吸附物弛豫。
进一步地,所述步骤s3中采用nudgedelasticband方法搜索过渡态,确定反应所需活化能e*。
进一步地,确定过渡态的具体方法为,使用materialstudio的castep模块搜索过渡态,并对过渡态的结构进行频率分析,如果只有一个虚频则表明搜索到的结构是反应的过渡态。
进一步地,所述步骤s4中通过对比分析不同的掺杂金属对反应能和活化能的影响,得出9种viii族金属对甲烷分解促进作用的大小,进而筛选比较合适的掺杂金属。
本发明的有益效果为:1.通过仿真模拟和数据分析得出,在cu(111)表面掺杂的9种viii族金属都能降低甲烷分解的活化能,其中,ru和rh掺杂的cu(111)表面对甲烷分解反应最有利。
2.通过仿真模拟发现,甲烷分解的基元反应符合brøsted–evans–polanyi原则,即反应的活化能和反应能呈线性关系,可以根据本发明绘制的线性关系图预测其它掺杂金属对甲烷分解反应的影响,为实验研究提供理论基础,筛选出合适的催化剂后再进行实验研究,更加省时省力,节约能源。
附图说明
图1:本发明铜表面掺杂viii族金属促进甲烷分解的模拟仿真方法的步骤图;
图2:本发明cu(111)表面和viii族金属掺杂的cu(111)表面模型结构的俯视图;
图3:本发明ni-cu表面甲烷分解过程各基元反应的始态、过渡态、终态结构示意图;
图4:本发明10种模型表面甲烷分解反应的反应能和活化能数据图;
图5:本发明10中模型表面甲烷分解反应活化能对比图;
图6:本发明ru-cu、ni-cu、os-cu、pd-cu、cu表面甲烷分解反应路径势能图;
图7:本发明甲烷分解反应中活化能和反应能的线性关系图。
具体实施方式
下面结合附图和具体实施例对本发明作进一步的详细描述。
本发明中铜表面掺杂viii族金属促进甲烷分解的模拟仿真方法的过程如图1所示:首先,建立cu(111)表面和viii族金属掺杂的cu(111)表面的共10种表面模型,其中,viii族金属掺杂方式为替代cu(111)表面第一层的1个cu原子;其次,根据密度泛函理论,优化cu(111)表面和viii族过渡金属掺杂的cu(111)表面甲烷分解反应始态和终态的结构,确定反应能e;然后根据反应始态和终态的结构搜寻反应过渡态的结构并确定反应活化能e*;最后分析不同掺杂元素对甲烷分解反应的影响,筛选比较合适的掺杂元素。
本实施例采用模拟仿真软件对ni掺杂的cu(111)表面甲烷分解的过程进行仿真模拟。
首先需要建立cu(111)表面的模型,cu的晶胞结构为面心立方晶胞,将其导入materialstudio模拟软件后沿(111)方向切面,原子层厚度设置为4层,选取p(3×3)超晶胞构建模型,设置真空层厚度为15å即得到cu(111)表面的模型图,如图2a所示为cu(111)模型的俯视图,把cu(111)模型的第一层的一个cu原子(图2b中白色圆球)替换为ni原子(图2b中黑色圆球)即可得到ni-cu表面的模型。
催化剂模型构建完成后需要确定各基元反应始态和终态最稳定的吸附构型,甲烷的基元包括分为ch4→ch3+h,ch3→ch2+h,ch2→ch+h,ch→c+h四步。因此,需要确定ch4、ch3、ch2和ch在催化剂表面最稳定的吸附构型,作为各基元反应的始态;确定ch3与h共吸附、ch2与h共吸附、ch与h共吸附、c与h共吸附时的最稳定构型,作为各基元反应的终态。图3a左侧图形为ch4在ni-cu表面的吸附构型,ch4的一个h原子朝下指向ni-cu表面的ni原子,该h原子与ni的距离为2.53å,ch4中c原子与ni的距离为3.61å,ch4在ni-cu表面吸附较弱,是一种物理吸附状态。图3b、c、d左侧图形分别为ch3、ch2、ch在ni-cu表面的吸附构型,三者最稳定的吸附位均为由一个ni原子和两个cu原子组成的中空位。在ch3吸附构型中,c原子到ni原子的距离为2.13å;在ch2吸附构型中,c原子到ni原子的距离为1.93å;在ch吸附构型中,c原子到ni原子的距离为1.85å。由吸附构型可见,由于ni的掺杂,使得反应始态各吸附物更容易吸附在ni的附近,并且掺杂ni后能使吸附更加稳定。图3a、b、c、d右侧图形分别为ch3与h共吸附、ch2与h共吸附、ch与h共吸附、c与h共吸附时的最稳定构型,共吸附的h原子均吸附在与chx(x=0-3)相对的中空位。优化完始态和终态的结构,由基元反应终态的能量减去始态的能量即可得到基元反应的反应能e,如图4所示,ni-cu表面ch4→ch3+h,ch3→ch2+h,ch2→ch+h,ch→c+h四步反应的反应能e分别为0.26ev、0.29ev、0.11ev、1.04ev,表明四个基元步骤均为吸热过程。
在上述结构优化过程中参数设置为,电子与电子之间的交换关联能均采用gga泛函数进行校正,平面波截断能取400ev,布里渊区积分k点取值3×3×1,电子自洽迭代和离子弛豫的收敛标准分别设置为能量收敛至10-5ev和原子力收敛至0.02ev/å。在结构优化过程中,ni-cu模型下部两层原子固定,顶部两层原子及吸附物弛豫。
有了基元反应始态和终态的结构后就可以搜索过渡态,本发明使用materialstudio的castep模块,采用nudgedelasticband方法搜索过渡态,得到过渡态的结构并对过渡态的结构进行频率分析,如果只有一个虚频则表明搜索到的结构是反应的过渡态。图3a、b、c、d中间的图形为各基元反应的过渡态结构,在每个过渡态结构中,均有一个h原子与c原子断开,断开的h原子吸附在ni或者cu的顶部,断开的h原子到c原子的距离分别为1.62å、1.91å、1.85å、1.79å。用过渡态的能量减去始态的能量得到活化能e*,如图4所述,ni-cu表面四个基元反应的活化能e*分别为0.85ev、0.54ev、0.43ev、1.35ev,ch→c+h反应所需活化能最高,为甲烷分解过程的速率控制步骤。
用与上述实施例同样的模拟方法,得到fe-cu、co-cu、ru-cu、rh-cu、pd-cu、pt-cu、os-cu、ir-cu、cu表面甲烷分解过程的结构图,并计算出反应能和活化能,反应能和活化能数据汇总如图4所示。对比不同催化剂模型表面的数据可以看出,相比于cu表面,掺杂viii族金属后四个基元反应的活化能均降低,不同的掺杂金属对活化能的降低程度不同。ru-cu和rh-cu表面,ch2→ch+h的反应能为负值,表明该反应为放热过程,其它反应能均为正值即为吸热过程。在10种催化剂模型表面,ch→c+h始终是活化能最高的一步基元反应,为甲烷分解过程的速率就控制步骤。说明掺杂的金属并没有改变反应的速率控制步骤,但是掺杂的金属能促进c-h键的断裂,降低反应的活化能,降低反应所需的温度,节约能源。
为了比较不同掺杂金属对活化能降低程度的影响。绘制如图5所示的活化能对比图,从ir-cu开始,各基元反应的活化能有明显上升,表明ru、rh、fe、co、ni掺杂后的表面对甲烷分解反应的促进作用要优于os、ir、pt、pd掺杂的表面。此外,ch→c+h反应在fe-cu表面活化能最低,这也解释了一些实验研究中fe催化剂表面容易发生积碳的原因,fe的掺杂使得ch分解得到c更加容易,会导致c在催化剂表面积累,导致催化剂烧结或失活。因此要筛选合适的掺杂金属,活化能降低太少不能有效促进甲烷分解,活化能降低太多又容易发生积碳导致催化剂失活。
为了更加直观的分析掺杂金属对甲烷分解在热力学和动力学上的影响,绘制了如图6所示的反应路径势能图。由于ru和rh性质相似,fe、co、ni性质相似,os和ir对甲烷分解的影响相似,pd和pt性质相似,选取了ru-cu、ni-cu、os-cu、pd-cu、cu共5种模型表面的数据绘制了反应路径势能图。图中ts1、ts2、ts3、ts4分别表示4步基元反应的过渡态,相比于cu表面,掺杂的金属使得甲烷分解反应在热力学和动力学上均更加有力,其中ru掺杂效果最好,在动力学上,4个基元反应的活化能相比于cu模型表面分别降低了0.94ev、0.81ev、0.60ev、0.56ev,ru的掺杂使反应在热力学也更加容易进行,尤其ch2→c+h在ru-cu表面反应能为-0.02ev,变为放热过程。ru和rh的掺杂对甲烷分解反应的影响是相似的,根据模拟仿真结果,在viii族金属中,ru或rh作为掺杂金属掺杂在cu(111)表面对甲烷分解反应最有利,这个结论可以更好地指导实验研究,合成高效的催化剂,促进甲烷转化成高价值化学品。
根据图4中的数据,以每个基元反应的反应能为横坐标,活化能为纵坐标可以绘制出反应能和活化能的关系图。如图7所示,甲烷分解反应的每个基元步骤均符合brøsted–evans–polanyi原则,即基元反应的活化能和反应能呈线性关系,反应的动力学是受热力学控制的,热力学上越有利,则反应动力学上越有利,反应越快。ch4→ch3+h,ch3→ch2+h,ch2→ch+h,ch→c+h四个基元步骤线性关系图的斜率分别为1.22、1.28、0.92、0.88。在所述整个模拟仿真过程中,搜索过渡态的过程比较繁琐,最耗时间,有了线性关系图,可以只优化基元反应的始态和终态结构,把计算出的反应能带入线性关系式估算反应活化能,可以将该线性关系图应用在除本发明所研究的9种掺杂金属以外,这样在筛选其他掺杂金属时就可以节约很多时间。
综合以上模拟仿真过程可以得出以下结论,在fe、co、ni、ru、rh、pd、pt、os、ir这9种viii族金属中,ru、rh作为掺杂金属掺杂在cu(111)表面对甲烷分解反应最有利,ru或rh的掺杂使得ch4→ch3+h,ch3→ch2+h,ch2→ch+h,ch→c+h的活化能降幅均超过0.5ev,意味着基元反应能在更低的温度下越过活化能能垒,使反应顺利进行,降低反应温度,节约能源。模拟仿真过程得到的如图7所示的活化能和反应能的线性关系图可以应用于本发明所研究的9种掺杂金属以外的元素,由反应能的数据预测活化能,节约模拟仿真过程所需的时间,更加快速地筛选出合适的催化剂,为实验研究提供理论指导,避免在实验过程中做很多无用功,有利于提高科研效率。
- 还没有人留言评论。精彩留言会获得点赞!