基于二级结构和多模型融合的非编码RNA与蛋白质相互作用预测方法与流程
基于二级结构和多模型融合的非编码rna与蛋白质相互作用预测方法
技术领域
[0001]
本发明属于生物信息学与计算机应用领域,尤其涉及一种基于二级结构和多模型融合的非编码rna与蛋白质相互作用预测方法。
背景技术:
[0002]
非编码rna参与很多复杂的细胞活动进程,在选择性剪切、染色质修饰和表观遗传等生命过程中发挥着重要作用,并与许多疾病有着密切的联系。研究表明,大部分非编码rna通过与蛋白质相互作用实现其调控功能。因此,研究非编码rna与蛋白质相互作用对于揭示非编码rna在人类疾病和生命活动中的分子作用机制具有重要意义,已成为当下分析非编码rna和蛋白质功能的重要途径之一。clip-seq,rip-seq等一些实验技术已经开发用于揭示非编码rna与蛋白质的相互作用,但是耗时长而且成本昂贵。所以,建立一个可靠准确的计算预测模型是非常重要的。
[0003]
近年来,一些计算方法被提出用于非编码rna与蛋白质相互作用预测。例如,catrapid利用氢键、范德华力等理化性质对非编码rna-蛋白质对进行评分。lncpro进一步使用fisher线性判别分析来提高预测性能。这两种方法采用手工构造特征的方式,需要较强的生物领域知识。随着机器学习在各个领域的广泛使用并展现出了良好的分类性能,许多科研人员提出了基于机器学习的非编码rna与蛋白质预测方法。rpiseq使用k-mer频率特征表示方法提取了rna和蛋白质序列的3-mer和4-mer频率特征,并分别训练了随机森林(rf)和支持向量机(svm)分类器。后来,cfrp通过非线性运算从rna和蛋白质的k-mer特征中提取出复杂特征,然后通过随机森林的特征重要性排序方法筛选关键特征,最终使用随机森林作为分类器,并产生了比较优异的分类结果。以上研究表明,机器学习方法的兴起对非编码rna与蛋白质相互作用预测起到了推进作用,提升了预测的准确性。另外,上述方法在两个主要方面存在局限性,首先,序列编码方法仍有待改进,其广泛使用的k-mer频率特征表示方法较为单一,不能全面地表征rna和蛋白质。而且,提取较短序列的3-mer和4-mer特征向量比较稀疏,丢失了序列的一些重要信息,不利于对机器学习模型进行训练,从而无法获得理想的分类性能。另外,随着深度学习在各个领域的应用,如图像分类,语义识别,并展现出了优异的性能。目前,越来越多的深度学习算法被开发应用于生物信息学领域。
技术实现要素:
[0004]
为了解决上述现有技术中存在的问题,本发明提供一种基于二级结构和多模型融合的非编码rna与蛋白质相互作用预测方法,该方法采用基于高阶序列-结构联合特征构造方法,将非编码rna和蛋白质的序列和二级结构联系起来,作为深度学习和机器学习融合模型的输入,在基准数据集上对提出的方法进行了评价,该方法能在不丢失关键rna和蛋白质序列信息的情况下,准确高效地预测出给定非编码rna与蛋白质是否相互作用,实验结果显示,本方法取得了比传统方法更高的分类性能。
[0005]
技术方案如下:
[0006]
一种基于二级结构和多模型融合的非编码rna与蛋白质相互作用预测方法,步骤如下:
[0007]
s1:整理实验所需数据集,获取rna和蛋白质的二级结构:
[0008]
s11:获取现有基准数据集;
[0009]
s12:使用rna和蛋白质的二级结构预测工具获取数据集中序列的二级结构序列;
[0010]
s2:构造rna和蛋白质的高阶序列-二级结构联合特征:
[0011]
根据序列-二级结构联合特征构造方法表征序列与其二级结构之间的联系,采用高阶编码策略来构造特征;
[0012]
s3:构建非编码rna与蛋白质相互作用预测模型,训练并对模型进行评估:
[0013]
在步骤s2的基础上,构建基于高阶序列-二级结构联合特征的机器学习与深度学习融合预测模型,然后使用整理后的数据集对模型进行训练和评估。
[0014]
进一步的,步骤s3预测模型构建的具体步骤包括:
[0015]
s31:搭建卷积神经网络模型,包括rna子cnn模型和蛋白质子cnn模型,然后通过全连接层对两个子模型进行输出联合,使用步骤s2提取出的特征对该深度学习模型进行训练;
[0016]
s32:建立随机森林机器学习模型,提取步骤s31训练后的cnn模型的全连接层输出特征,训练该随机森林模型;
[0017]
s33:使用基准数据集对模型进行评估,并与现有的相关方法进行比较。
[0018]
进一步的,步骤s33中,评估指标包括:准确性、敏感性、特异性、精密度、马修斯相关系数和roc曲线下面积,计算公式如下:
[0019][0020][0021][0022][0023][0024]
其中:accuracy表示准确性、sensitivity表示敏感性、specificity表示特异性、prediction表示精密度、mcc表示马修斯相关系数,tp表示预测正确的正样本数、tn表示预测正确的负样本数,fp表示预测错误的正样本数,fn表示预测错误的负样本数。
[0025]
本发明的有益效果是:
[0026]
本发明所述的基于二级结构和多模型融合的非编码rna与蛋白质相互作用预测方法具有以下有益效果:
[0027]
1、本发明的模型不同于以往对序列使用简单的k-mer编码特征表示方法,克服了以往特征中丢失了一些重要信息导致分类性能差的缺陷;设计了一种高阶序列-二级结构
特征构造方法来充分发掘非编码rna和蛋白质序列与二级结构之间的联系,并且考虑了邻接核苷酸和邻接氨基酸之间的依赖性。并且该方法为提取不等长序列的特征提供了新的思路。
[0028]
2、为了充分挖掘rna和蛋白质之间的联系,本发明使用cnn模型来学习特征之间的局部信息,并且在cnn模型中,使用全连接层将rna和蛋白质的高水平特征联合,并对高效的随机森林模型进行训练。
附图说明
[0029]
构成本申请的一部分的附图用来提供对本发明的进一步理解,本发明的示意性实例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
[0030]
图1为本发明高阶序列-二级结构联合特征构造方法的示意图;
[0031]
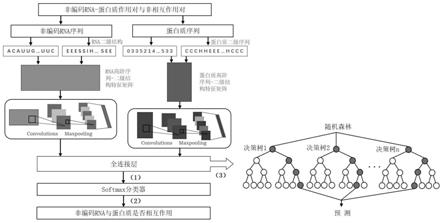
图2为本发明预测模型的结构图。
具体实施方式
[0032]
下面结合附图1-2对基于二级结构和多模型融合的非编码rna与蛋白质相互作用预测方法的实施例进行详细说明,但是本发明可以由权利要求限定和覆盖的多种不同方式实施。
[0033]
实施例1
[0034]
s1:整理实验所需数据集,获取rna和蛋白质的二级结构。
[0035]
对于分类问题,建立合理的基准数据集是预测高精度分类效果的必要步骤。正、负样本分布、数据集大小和噪声与深度学习的学习质量密切相关。同时,根据之前的研究显示,rna和蛋白质的结构决定其生物学功能,因此通过引入非编码rna和蛋白质的二级结构可以更好地表征rna和蛋白质,提升预测的准确性。
[0036]
进一步地,步骤s1数据集构造的具体步骤包括:
[0037]
s11:在我们的工作中,所有的数据集来自先前研究(ipminer,rpiseq,rpi-pred)通过实验验证所得到的基准数据集。
[0038]
s12:使用rna和蛋白质的二级结构预测工具获取数据集中序列的二级结构序列。
[0039]
s2:构造rna和蛋白质的高阶序列-二级结构联合特征。
[0040]
传统的简单k-mer编码方法忽视了rna和蛋白质的序列与其二级结构之间的联系,丢失了一些重要信息,导致后续机器学习模型的预测结果不理想。因此,我们采用序列-二级结构联合特征构造方法表征序列与其二级结构之间的联系,并且考虑到邻接核苷酸和邻接氨基酸之间的依赖性,我们采用了高阶编码的策略来构造特征。
[0041]
s3:构建非编码rna与蛋白质相互作用预测模型,训练并对模型进行评估。
[0042]
在步骤s2的基础上,构建基于高阶序列-二级结构联合特征的机器学习与深度学习融合预测模型,然后使用整理后的数据集对模型进行训练和评估。
[0043]
进一步地,步骤s3预测模型构建的具体步骤包括:
[0044]
s31:搭建卷积神经网络(cnn)模型,包括rna子cnn模型和蛋白质子cnn模型,然后通过全连接层对两个子模型的输出联合,使用步骤s2提取出的特征对该深度学习模型进行训练。
[0045]
s32:建立随机森林机器学习模型,提取步骤s31训练后的cnn模型的全连接层输出特征,训练该随机森林模型。
[0046]
s33:使用基准数据集对模型进行评估,并与现有的相关方法进行比较。
[0047]
实施例2
[0048]
如图1-2所示,本实例公开一种基于高阶序列-二级结构联合特征表示方法的非编码rna与蛋白质相互作用的预测方法,其过程是基于python3.6-tensorflow1.5-keras2.2-scikit-learn0.23.0,包括以下步骤:
[0049]
s1:整理实验所需数据集,获取rna和蛋白质的二级结构。
[0050]
对于分类问题,建立合理的基准数据集是预测高精度分类效果的必要步骤。正、负样本分布、数据集大小和噪声与深度学习的学习质量密切相关。同时,根据之前的研究显示,rna和蛋白质的结构决定其生物学功能,因此通过引入非编码rna和蛋白质的二级结构可以更好地表征rna和蛋白质,提升预测的准确性。
[0051]
s2:构造rna和蛋白质的高阶序列-二级结构联合特征。
[0052]
采用序列-二级结构联合特征构造方法表征序列与其二级结构之间的联系,并且考虑到邻接核苷酸和邻接氨基酸之间的依赖性,我们采用了高阶编码的策略来构造特征。以rna为例,如图1所示,建立rna二阶序列-二级结构联合特征矩阵,将矩阵中的所有元素初始化为0。由于rna序列的氨基酸种类数为4,二级结构的种类数为6,所以特征矩阵大小为16
×
64。然后使用长度k=2的滑动窗口同时遍历rna序列和对应的二级结构序列,每滑动一次,矩阵的对应位置的元素加1。
[0053]
s3:构建非编码rna与蛋白质相互作用预测模型,训练并对模型进行评估。
[0054]
在步骤s2的基础上,构建基于高阶序列-二级结构联合特征的机器学习与深度学习融合预测模型,如图2所示。然后,使用整理后的数据集对模型进行训练和评估。
[0055]
进一步地,在本实施例中,步骤s1具体包括以下步骤:
[0056]
s11:在我们的工作中,实验所需的数据集来自先前的ipminer,rpiseq,rpi-pred研究。具体信息见表1。为了使得数据集的正、负样本均衡,随机构造了与非编码rna-蛋白质作用对数量相当的非作用对,这些非作用对未出现在非编码rna与蛋白质相互作用的数据集中。
[0057]
表1本发明所使用的数据集详情
[0058][0059]
s12:在步骤s11的基础上,对于非编码rna,给定一个非编码rna序列,序列由a,u,g,c组成,我们首先使用viennarnapackage2.0工具包中的rnafold预测出该序列的以点括号组成的二级结构序列,然后以点括号表示的非编码rna二级结构序列作为输入,使用bprna预测出由7种rna二级结构组成的非编码rna二级结构序列,这7种结构和对应的表示字符分别为茎(s)、发卡环(h)、多分支环(m)、内环(i)、突环(b)、外环(x)和未配对单链(e)。
由于外环与多分支环结构相似,所以在这里我们将外环视为多分支环。对于蛋白质,给定一个蛋白质序列,序列由20种氨基酸组成,我们使用蛋白质二级结构预测工具scartch获取其对应的二级结构序列,由3种结构组成,分别为
ɑ
螺旋(h)、β折叠(e)和无规则卷曲(c)。根据理化性质,我们将20种氨基酸分成7组,并编号为0-6。
[0060]
在本实施例中,步骤s3具体包括以下步骤:
[0061]
s31:为了充分挖掘rna、蛋白质的序列和二级结构的内部联系,我们搭建两个子卷积神经网络(cnn)模型学习提取后的特征,然后使用全连接层对cnn处理后的高水平特征进行联合,然后输入到softmax分类器进行相互作用预测。
[0062]
s32:抽取cnn模型训练后的全连接层中的非编码rna-蛋白质联合高水平特征向量,同时构建随机森林机器学习模型,并使用这些高水平特征向量对随机模型进行训练。
[0063]
s33:在模型训练好之后,我们使用基准数据集对我们发明的基于高阶序列-二级结构联合特征构造方法的深度学习与机器学习融合预测模型(rpi-cr)进行了评估,本发明采用多种评价指标对预测非编码rna与蛋白质相互作用的结果性能进行评价,包括准确性(accuracy)、敏感性(sensitivity)、特异性(specificity)、精密度(prediction)、马修斯相关系数(mcc)和roc曲线下面积(auc)。这些指标的计算公式如下:
[0064][0065][0066][0067][0068][0069]
其中,tp、tn分别代表预测正确的正样本数和负样本数,fp、fn分别代表预测错误的正样本数和负样本数,这些指标被广泛地应用于评价该领域模型的性能。并且,我们使用指标sum(以上6个指标之和)对这些方法进行综合评价。表2列出了本方法与现有相关方法的性能指标比较情况,从表中可以看出,本发明虽然在几个指标上不如其他方法,但在总体上优于lncpro,rpiseq和cfrp。
[0070]
表2本发明与其他方法在基准数据集上的比较
[0071][0072]
结果表明,我们的方法通过引入非编码rna和蛋白质二级结构以及序列和二级结构联合高阶特征构造方法,对于非编码rna和蛋白质的相互作用预测是有帮助的、起作用的。同时,我们的方法可以扩展到解决其他序列相关的预测问题研究中。
[0073]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1