一种天名精提取物及制备方法和在通过JAK2/STAT3通道抗肝癌活性药物中的应用与流程
一种天名精提取物及制备方法和在通过jak2/stat3通道抗肝癌活性药物中的应用
技术领域
[0001]
本发明属于中药技术领域,特别涉及一种天名精提取物及制备方法和在通过jak2/stat3通道抗肝癌活性药物中的应用。
背景技术:
[0002]
天名精属是菊科的一个属,约有21种,主要分布于欧洲南部和亚洲。其中,在中国西南山区有17种。天名精作为一著名的民族药,在土家族、白族、苗族等少数民族都有着悠久的药用历史。研究发现其主要含有倍半萜内酯等化学成分,且大部分含有α-亚甲基-γ-丁内酯,具有抗癌、抗炎、抗菌等生物活性。前期通过cck-8活性筛选发现倍半萜内酯总提取物(slec)对hepg-2细胞有较好的作用(ic
50
=4.2μg/ml),但其有效成分及抗肝癌作用机制都尚不清楚。
[0003]
现代中医药研究的目标是阐述中药哪些化学成分有效,以及是如何起作用的。虽然传统的提取、分离和分析方法已经形成了一个非常完整的体系,但仍存在耗时、费力、周期长等缺点,不能满足和适应现代中医药研究。
[0004]
其中,高速逆流色谱(hsccc)是一种非常有效的应用于中药分离纯化的工具。与其他分离方法相比,它不是使用固体载体作为固定相,而是将分离的样品分散在不能混溶的两相液体中,以达到分离的效果。因此,hsccc具有装载能力高、制备效率高、溶剂消耗低、重复性好等优点。
[0005]
高效液相色谱串联四极杆飞行时间质谱(hplc-q-tof-ms
e
)可以快速获取精确的分子离子峰和碎片离子等信息,然后分析并鉴定不同类型的化学成分。因此,我们采用hplc-q-tof-ms
e
策略对天名精中的倍半萜类成分进行鉴定,并指导hsccc快速分离出活性部位的量大化合物,最后通过jak2/stat3信号通路将分离出的化合物用于抗肝癌活性。
技术实现要素:
[0006]
为了克服现有技术中存在的缺点与不足,本发明的首要目的在于提供一种天名精提取物的制备方法。
[0007]
本发明的又一目的在于提供一种上述制备方法制备得到的天名精提取物。
[0008]
本发明的再一目的在于提供一种上述制备方法制备得到的天名精提取物的应用。
[0009]
本发明的目的通过下述技术方案实现:
[0010]
一种天名精提取物的制备方法,包括以下操作步骤:
[0011]
(1)取天名精全草,干燥粉碎,用天名精全草10倍质量的体积百分比浓度95%的乙醇浸泡提取24h,提取三次,将三次提取液混合,浓缩,干燥后得到总提取物;向总提取物添加质量百分比浓度2%的naoh溶液进行溶解,直至ph=9,停止添加naoh溶液,然后用质量百分比浓度1%的浓盐酸调整ph至2,得到沉淀,得到天名精总提取物;
[0012]
(2)将天名精总提取物采用高速逆流色谱法分离获得三个化合物,分别是结构式
为c
15
h
22
o4的化合物1,结构式为c
15
h
20
o3的化合物2,结构式为c
15
h
20
o3的化合物3;
[0013]
所述高速逆流色谱法的参数为:先采用流速为3.0ml/min、转速为835rpm、采样量为200mg,体积比1:9:9:1的正己烷-乙酸乙酯-甲醇-水溶剂体系运行150min时,泵停止工作;然后将溶剂体系替换为体积比4:3:2的氯仿-甲醇-水;运行320min后,关闭泵,从而在第一阶段的60-66min得到a峰、在74-82min得到b峰、在第二阶段的80-92min得到c峰,分别对应于化合物1、化合物2和化合物3;
[0014]
一种由上述的制备方法制备得到的天名精总提取物。
[0015]
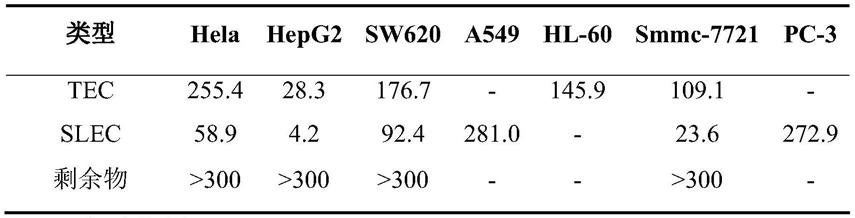
上述的天名精总提取物在通过jak2/stat3通道抗肝癌活性药物中的应用。
[0016]
化合物1在通过jak2/stat3通道抗肝癌活性药物中的应用。
[0017]
化合物2在通过jak2/stat3通道抗肝癌活性药物中的应用。
[0018]
化合物3在通过jak2/stat3通道抗肝癌活性药物中的应用。
[0019]
本发明相对于现有技术具有如下的优点及效果:
[0020]
(1)本发明首次采用碱溶酸沉的方法对天名精的总倍半萜成分进行提取;
[0021]
(2)本发明采用了高速逆流色谱分离出天名精提取物中含量最高的三个活性物质,克服了现有技术天名精提取工艺耗时、费力的缺陷;
[0022]
(3)本发明提取得到的天名精提取物具有非常显著的通过jak2/stat3通道抗肝癌活性。
附图说明
[0023]
图1为slec部位的正、负tic图和uv图。
[0024]
图2和图3为hsccc制备化合物的色谱图。
[0025]
图4为化合物1-3对hepg2细胞中jak2和stat3 mrna表达的影响。
[0026]
图5为化合物1-3对hepg2细胞中jak2、p-jak2、stat3、p-stat3蛋白表达的影响。
具体实施方式
[0027]
下面结合具体实施例及附图进一步说明本发明的内容,但不应理解为对本发明的限制。若未特别指明,实施例中所用的技术手段为本领域技术人员所熟知的常规手段。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
[0028]
实施例1:
[0029]
一、本实施例所使用的方法:
[0030]
1、天名精提取物的制备
[0031]
取天名精全草(2kg),干燥粉碎,用天名精全草10倍质量的体积百分比浓度为95%的乙醇浸泡24h,提取三次,将三次提取液混合,干燥后得到总提取物(tec)458g。将tec(400g)加质量百分比浓度2%的naoh溶液进行溶解,直至ph=9,然后用质量百分比浓度1%的浓盐酸调整ph至2,得到沉淀,最终得到总倍半萜内酯总提取物(slec)(42g)。
[0032]
2、cck-8法筛选抗肿瘤活性
[0033]
取对数生长期的细胞,消化计数,以3*103个细胞/孔密度接种于96孔板内,每孔100μl。各组均设3个复孔,培养贴壁后将不同浓度(50、100、150、200、250g/ml)的tec部位、slec部位和剩余物样品,分别进行加药处理相应的时间后,按照每孔加入10μl/孔的cck8,
用完全培养基配置cck8溶液(按照1ml完全培养基加入100ul的cck8溶液),去除含药培养基每孔加入100ul含有cck8的培养基。37℃,5%co2继续孵育4小时后于bio-tek酶标仪分析450nm处吸光度(od)值。
[0034]
3、活性部位的液质分析
[0035]
采用hplc-q-tof-ms
e
对slec部位的化学成分进行分析。液相条件为:安捷伦高效液相色谱柱(eclipse xdb c
18
,4.6
×
250
×
5μm);流动相a为0.1%甲酸水,流动相b为乙腈,洗脱程序:0-8min,2%b;8-10min,2%-20%b;10-50min,20%-52%b;50-60min,52%b;60-65min,52%-80%b;65-68min,80-98%b;68-75min,98%b柱温25℃,体积流量1.0ml/min,注入体积5μl。
[0036]
质谱条件为:质谱条件离子化方式电喷雾离子化(esi),正、负离子模式分别采集,干燥气温度325℃,干燥气体积流量为6.8l
·
min-1,鞘气温度350℃,毛细管电压4.0kv,碎片电压130v,质量数范围为m/z 50~1000。
[0037]
4、hsccc的制备方法
[0038]
4.1分配系数的测定
[0039]
本实验采用hplc法计算slec部位在不同溶剂系统中的分配系数。具体操作为:按溶剂体系比例配制一定体积(20ml)的上下相溶剂,待分相平衡后,各取相同体积的上下相溶剂放置同一容器中(10ml试管),加入一定量(0.1g)的样品,剧烈震摇使溶解,静置。取相同体积的上下相溶剂,注入hplc测定目标成分在上下相中的峰面积as和am,根据as/am计算分配系数k值。
[0040]
4.2hsccc分离
[0041]
溶剂系统主要根据目标物的分配系数进行选择。五个溶剂系统:正己烷-乙酸乙酯-甲醇-水(1:1:1:1)、正己烷-乙酸乙酯-甲醇-水(1:9:9:1)、正己烷-乙酸乙酯-甲醇-水(3:1:1:1)、正己烷-甲醇-水(5:4:1)和氯仿-甲醇-水(4:3:2)分别用于进行slec部位分离实验。
[0042]
每次分离实验时,以30.0ml/min的固定相填充分离柱,然后设正转(fwd),转速为835rpm。流动相以3.0ml/min的速度泵入仪器中。达到平衡后,将样品注射到注射阀中。在218nm处连续监测,用紫外检测器监测峰组分。
[0043]
5、体外细胞实验
[0044]
用cck-8法检测分离的化合物(1-3)对hegg-2细胞株的细胞毒性。
[0045]
5.1定量实时聚合酶链反应
[0046]
使用trizol试剂从hepg-2细胞中提取总rna。使用primescript ii rtase逆转录酶对每个样本进行大约500ng的总rna进行反向转录,以制备cdna。定量实时聚合酶链反应(qrt-pcr)使用cfx-connect 96和sybr快速qpcr主混料进行。pcr引物序列如下:jak2,正向:actaaatgctgtccccc,反向:ttcatgcagttgaccgt;stat3,正向:tgagggagcagagatgtg,反向:tgggggcttggtaaaa;gapdh,正向:ccactcctccacctttg,反向:caccaccctgttgctgt。每个目标基因的相对gapdh表达计算使用2-δδc
方法。
[0047]
5.2免疫印迹检测
[0048]
使用ripa裂解缓冲液从ra-fls中提取总蛋白,用bca蛋白检测试剂盒检测蛋白浓度。待20mg的样品用200mol/l裂解缓冲液完全裂解后,4℃继续1200rpm离心20min,取上清
进行western blot分析。每个样品的蛋白浓度由bicinchoninic acid protein assay kit测定。在10%的sds-聚丙烯酰胺凝胶(sds-page)上电泳并分离负载蛋白,然后转移到聚偏二氟乙烯(pvdf)膜上。室温下将pvdf膜在5%脱脂牛奶中封闭1小时,与一抗(jak2、anti-jak2、stat3、anti-stat3、gapdh)在4℃条件下孵育过夜。pbst清洗膜4次,每次5分钟。加入山羊抗兔igg(1:10000),室温孵育1h,ecl检测,通过tanon gis软件读取相关条带灰度值。
[0049]
6、统计分析
[0050]
以标准差(sd)表示,组间比较采用单因素方差分析(anova)和t检验。所有统计分析均采用spss 15.0在p<0.05或p<0.01处具有显著性。
[0051]
二、实验结论
[0052]
1、cck-8法对tec和slec部位的抗肿瘤作用
[0053]
采用cck8法评价tec和slec部位对肿瘤细胞系hela、hepg2、sw620、a549、hl-60、smmc-7721和pc-3的细胞毒性。结果如表1所示。从表中可得,tec和slec对不同类型的肿瘤细胞均有一定的作用。尤其slec部位对hepg-2和smmc-7721细胞表现出良好的活性。
[0054]
表1:天名精各部位对各个癌细胞的ic
50
值
[0055][0056]
2、液质分析
[0057]
基于slec部位对肝癌细胞有较好的活性,因此采用hplc-q-tof-ms/ms对其化学成分进行分析。通过液相和质谱条件优化,分别在正、负模式下进行质谱分析,如图1和表2所示。研究发现这种类型的化合物(倍半萜内酯类)在正模式下具有更高的响应,主要以[m+h]
+
的形式存在,进一步研究分子离子峰和多级质谱的裂解规律,发现存在大量异构体。因此,具体的化合物名称还需要通过其他方法进一步确定。
[0058]
表2:slec部位的化学成分研究
[0059][0060][0061]
3、hsccc快速分离
[0062]
选择合适的两相溶剂体系是hsccc高效分离的关键,通常需要高效液相色谱检测目标成分的合适k值。一般来说,k值应该在0.5到2.0之间。
[0063]
hplc分析三种目标化合物的k值如表3所示。从表中可以看出,a峰和b峰非常适合于溶剂体系2。但峰c不适于此,适合溶剂体系5。因此,当流动相运行150min时,泵停止工作,然后将溶剂体系替换为氯仿-甲醇-水(v/v/v,4:3:2)。320min后,得到a峰(60-66min)、b峰(74-82min)、c峰(80-92min),如图3所示。
[0064]
表3:hsccc分离不同溶剂体系中slec化合物的k值
5),1.87(m,1h,h-4),2.04(m,1h,h-6),2.10(m,1h,h-2),2.43(m,1h,h-3),2.39(dd,j=9.4,4.2hz,1h,h-9),2.70(m,1h,h-7),4.21(ddd,j=12.0,9.2,3.1hz,1h,h-8),5.44(d,j=3.2hz,1h,h-13),6.11(d,j=3.5hz,1h,h-13).
13
c nmr(151mhz,cdcl3)δ:222.0(c-1),169.8(c-12),140.2(c-11),120.1(c-13),80.8(c-8),50.0(c-10),48.7(c-5),44.8(c-7),44.1(c-3),35.2(c-2),34.5(c-9),29.6(c-4),24.1(c-6),22.0(c-14),20.0(c-15).
[0072]
5、活性验证
[0073]
实验表明slec部位对hepg-2细胞有显著的抑制作用。因此,本实验继续对三种分离的化合物进行体外细胞实验验证。结果表明三种化合物均表现出一定的抑制作用(ic50=9.83、2.95、4.15μm)。
[0074]
5.1化合物对hepg-2细胞jak2和stat3mrna表达的影响
[0075]
如图4所示,与对照组对比,阳性对照紫杉醇的jak-2和stat-3的mrna表达水平都显著降低(p<0.01)。给与化合物干预后,化合物1和2的高、中浓度的jak-2mrna表达水平较对照组显著降低(p<0.01);化合物3的jak-2mrna表达水平较对照组显著降低(p<0.05),且具有浓度依赖性;化合物1,2和3的stat-3mrna表达水平较对照组降低(p<0.01),且具有浓度依赖性。
[0076]
5.2化合物对hepg-2细胞jak2和stat3 mrna表达的影响
[0077]
如图4所示,与对照组对比,阳性对照紫杉醇的jak2和stat3的mrna表达水平都显著降低(p<0.01)。给与化合物干预后,化合物1和2的高、中浓度的jak2 mrna表达水平较对照组显著降低(p<0.01);化合物3的jak2 mrna表达水平较对照组显著降低(p<0.05),且具有浓度依赖性;化合物1,2和3的stat3 mrna表达水平较对照组降低(p<0.01),且具有浓度依赖性。
[0078]
5.3化合物对hepg2细胞jak2、p-jak2、stat3、p-stat3蛋白表达的影响
[0079]
如图5所示,与对照组对比,阳性对照紫杉醇的p-jak2和p-stat3蛋白表达水平都显著降低(p<0.01)。给与化合物干预后,化合物1和3的高、中浓度的p-jak2和p-stat3蛋白表达水平较对照组显著降低(p<0.05或p<0.01);化合物2的高浓度的p-jak2蛋白表达水平显著降低(p<0.05),化合物1和3的高、中浓度的p-stat3蛋白表达水平显著降低(p<0.05或p<0.01)。这提示化合物1、2、3与阳性药紫杉醇均能显著抑制p-jak2和p-stat3蛋白的表达。紫杉醇、化合物1-3的jak2、和stat3蛋白表达水平均无统计意义。
[0080]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1