基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法与流程
1.本发明涉及赖氨酸乙酰化位点预测研究与分析领域,尤其涉及一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法。
背景技术:
2.赖氨酸乙酰化是一种保守的蛋白质翻译后修饰,与多种代谢疾病密切相关,因此,赖氨酸乙酰化位点的识别对于代谢疾病治疗的研究具有重要意义。蛋白质结构特性包含高度有用的结构信息,为蛋白质翻译后修饰的鉴定提供了有力的依据;特征学习过程中,不同层级特征间的信息存在互补,同时关注不同层级特征的信息能够有效提高特征质量。现有的深度学习方法采用蛋白质序列层面的信息作为输入,未考虑到蛋白质结构特性;特征提取时仅考虑了高层级特征,导致信息严重丢失,进而降低预测结果。
技术实现要素:
3.本发明的目的在于避免现有技术的不足之处而提供一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法。
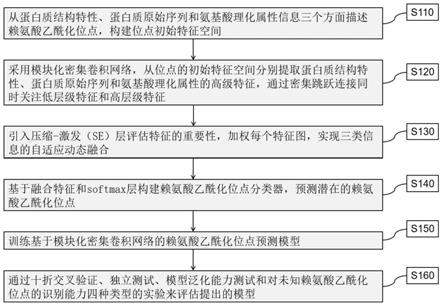
4.本发明的目的可以采用如下的技术措施来实现,设计一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法,包括:
5.从蛋白质结构特性、蛋白质原始序列和氨基酸理化属性信息三个方面描述赖氨酸乙酰化位点,构建位点初始特征空间;
6.采用模块化密集卷积网络,从位点的初始特征空间分别提取蛋白质结构特性、蛋白质原始序列和氨基酸理化属性的高级特征,通过密集跳跃连接同时关注低层级特征和高层级特征;
7.引入压缩-激发(se)层评估特征的重要性,加权每个特征图,实现三类信息的自适应动态融合;
8.基于融合特征和softmax层构建赖氨酸乙酰化位点分类器,预测潜在的赖氨酸乙酰化位点;
9.训练基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型;
10.通过十折交叉验证、独立测试、模型泛化能力测试和对未知赖氨酸乙酰化位点的识别能力四种类型的实验来评估提出的模型。
11.其中,从蛋白质结构特性、蛋白质原始序列和氨基酸理化属性信息三个方面描述赖氨酸乙酰化位点,构建位点初始特征空间的步骤包括:
12.(1)赖氨酸乙酰化位点的实验数据收集和预处理;
13.(2)通过编码方式将收集到的蛋白质数据转化为数值向量,构建位点初始特征空间,并作为预测模型的输入。
14.其中,赖氨酸乙酰化位点的实验数据收集和预处理,包括步骤:
15.从蛋白质赖氨酸修饰数据库(plmd)收集并下载了6078条、3645条和1860条经实验
验证的人类、小家鼠和大肠杆菌赖氨酸乙酰化蛋白质数据。
16.考虑到spider3服务器无法处理含有非标准氨基酸的蛋白质序列,本发明手动删除了这些蛋白质序列。以人类这一物种为例,利用cd-hit工具进行序列去冗余避免序列同源性较大而造成模型的偏差,阈值设定为0.4,保留了4977条乙酰化蛋白质序列。本发明将过滤后的4977条乙酰化蛋白质序列随机选择10%(498条)构建独立测试数据集,剩余乙酰化蛋白质序列作为训练数据集,便于与其他赖氨酸乙酰化位点预测器进行比较。
17.其中,通过编码方式将收集到的蛋白质数据转化为数值向量,构建位点初始特征空间,并作为预测模型的输入,包括步骤:
18.(1)使用one-of-21编码位点的蛋白质原始序列信息,对于长度为l的基序,将得到l
×
21维的蛋白质原始序列信息的向量表示;
19.(2)采用atchley因子编码位点的氨基酸理化属性信息,每个氨基酸残基由5个atchley因子表示,对于长度为l的基序,将得到l
×
5维的氨基酸理化属性信息的向量表示;
20.(3)通过spider3获取蛋白质结构特性信息,包括3种属性中的8个指数,即二级结构:α螺旋p(h)、β链p(c)、γ环p(e),局部骨干扭转角:ψ、θ、τ,可及表面积:asa。对于长度为l的基序,将得到l
×
8维的蛋白质结构特性信息的向量表示。
21.其中,采用模块化密集卷积网络,从位点的初始特征空间分别提取蛋白质结构特性、蛋白质原始序列和氨基酸理化属性的高级特征,通过密集跳跃连接同时关注低层级特征和高层级特征,包括步骤:
22.(1)引入模块化网络结构的设计思想,构建结构、序列和理化三个信息模块;
23.(2)采用堆叠密集卷积块对每个模块进行高级特征的提取,通过密集跳跃连接同时考虑低层级和高层级特征,实现不同层级特征间的信息互补。
24.其中,引入模块化网络结构的设计思想,构建结构、序列和理化三个信息模块,包括步骤:
25.基于蛋白质结构特性、蛋白质原始序列和氨基酸理化属性分别构建了结构模块、序列模块和理化模块和三个特征提取子模块,各子模块间参数空间相互独立,有效避免了三类信息之间的串扰,提高特征的质量。
26.其中,采用堆叠密集卷积块对每个模块进行高级特征的提取,通过密集跳跃连接同时考虑低层级和高层级特征,实现不同层级特征间的信息互补,包括步骤:
27.由于结构模块、序列模块和理化模块网络结构相同,这里仅对序列模块进行说明:
28.(1)首先,序列模块接收长度为l的位点基序的one-of-21编码作为输入,然后通过一维卷积层生成蛋白质原始序列信息的低级特征图,如公式(1)所示。
29.x0=σ(i*w+b)
ꢀꢀꢀꢀꢀꢀ
(1)
30.其中,i为one-of-21编码向量。为权重矩阵,s为过滤器的大小(s=3),d是过滤器数量(d=96)。b为偏置项,σ为激活函数。x0为一维卷积层的输出,大小为l
×
d。
31.(2)采用密集卷积块提取蛋白质原始序列信息的高级特征表示,密集卷积过程如公式(2)所示。
32.x
l
=σ([x0;x1;...;x
l-1
]*w
′
+b
′
)
ꢀꢀꢀꢀ
(2)
[0033]
其中,x
l-1
为密集卷积块中第l-1个卷积层生成的特征图,[
·
]表示沿特征维串联。
为权重矩阵,d
′
为密集卷积块中1到l-1层卷积的过滤器总数,d
″
为密集卷积块中第l个卷积层过滤器数(d
″
=32)。b
′
为偏置项,σ为激活函数,x
l
表示密集卷积块中第l个卷积层生成的特征图。密集卷积块的输出为低级特征图x0与密集卷积块中每个卷积层生成的特征图x1,x2,...,x
l
的特征维串联,即[x0;x1;...;x
l
]。
[0034]
(3)采用过渡层将(2)得到的蛋白质原始序列信息的特征图再进行卷积运算和激活操作,过渡层过程如公式(3)所示。
[0035]
x=σ([x0;x1;...;x
l
]*w
″
+b
″
)
ꢀꢀꢀꢀ
(3)
[0036]
其中,为权重矩阵,s
′
为过滤器的大小(s
′
=1)。b
″
为偏置项,σ为激活函数,x为过渡层的输出。之后,对过渡层结果进行平均池化操作,以减小特征图维数,降低模型过拟合风险。
[0037]
(4)重复(2)、(3)步骤,构成堆叠密集卷积块。第四次的(2)步骤后不进行(3)步骤,而是使用全局平均池化替代。
[0038]
经过上述过程,序列模块提取到位点的蛋白质原始序列的高级特征x
(seq)
。
[0039]
类似,理化模块和结构模块也通过上述过程提取到位点的氨基酸理化性质和蛋白质结构特性的高级特征。
[0040]
其中,引入压缩-激发(se)层评估特征的重要性,加权每个特征图,实现三类信息的自适应动态融合包括步骤:
[0041]
(1)引入压缩-激发(se)层评估特征的重要性,加权每个特征图;
[0042]
(2)蛋白质结构特性、蛋白质原始序列和氨基酸理化属性三类信息的自适应动态融合。
[0043]
其中,引入压缩-激发(se)层评估特征的重要性,加权每个特征图,包括步骤:
[0044]
以序列模块为例进行说明:
[0045]
(1)压缩(squeeze):采用全局平均池化将序列模块提取到的高级特征x
(seq)
的全局空间信息压缩到通道描述符中,压缩过程如公式(4)所示。
[0046][0047]
其中,z
c
表示x
(seq)
的第c个特征图的通道统计信息,f
sq
(
·
)表示压缩操作,w和h分别表示特征图的宽和高。将x
(seq)
的每个特征图都计算统计信息后,得到x
(seq)
的通道描述符
[0048]
(2)激发(excitation):采用两个全连接层(fc)捕获x
(seq)
的通道依赖性,学习x
(seq)
中每个特征图的特异性权值,激发过程如公式(5)所示。
[0049]
s=f
ex
(z,w)=σ(w2*δ(w1*z))
ꢀꢀꢀꢀ
(5)
[0050]
其中,表示激发操作后学习到的x
(seq)
中每个特征图的特异性权值,f
ex
(
·
)表示激发操作。δ和σ分别表示两个全连接层的激活函数,前者为relu函数,此层为具有参数的维度约减层,其缩减比为r(r=16);后者为sigmoid函数,此层为具有参数的维度还原层,确保s的维度与特征x
(seq)
的通道数相同。
[0051]
(3)特征缩放(scale):通过以下激活缩放x
(seq)
得到se层的输出其中为的任意一个元素,的计算如公式(6)所示。
[0052][0053]
其中,表示将特征图上的每个值与权值sc相乘。
[0054]
类似,理化模块和结构模块也通过se层得到加权后的高级特征。
[0055]
其中,蛋白质结构特性、蛋白质原始序列和氨基酸理化属性三类信息的自适应动态融合,包括步骤:
[0056]
se层是基于全局平均池化和两个全连接层(fc)实现的,且结构模块、序列模块和理化模块的网络结构相同,都通过se层得到加权后的高级特征。然后,串联每个子模块的输出,得到用于分类的融合特征由于se层为不同的特征图进行加权,使得特征融合过程具有自适应动态特性。
[0057]
其中,基于融合特征和softmax层构建赖氨酸乙酰化位点分类器,预测潜在的赖氨酸乙酰化位点,包括步骤:
[0058]
softmax层接收高级特征作为输入,经过加权求和和激活操作后得到样本的预测类别,softmax层的前向传播过程如公式(7)所示。
[0059][0060]
其中,为权重矩阵,为偏置项。p(y=i|x)表示样本x预测为i∈{0,1}类的概率,概率最大所对应的类别即为softmax分类器的预测类别。
[0061]
其中,训练基于模块化密集卷积网络赖氨酸乙酰化位点预测模型,包括步骤:
[0062]
(1)采用交叉熵作为代价函数,以最小化训练误差:
[0063][0064]
其中,n为训练样本总数,y
j
为第j个输入基序的真实标签,x
j
为第j个输入基序。
[0065]
(2)在训练中采用l2正则化,以减轻过拟合的影响,模型的最终目标函数为:
[0066]
min
w
(l
c
+λ∑(||w||2)2)
ꢀꢀꢀꢀꢀꢀ
(9)
[0067]
其中,λ为正则化系数,||w||2为权重矩阵的l2范数。
[0068]
(3)采用adam优化器对目标函数进行优化,学习率和批处理分别设置为0.0001和1000。采用early stopping策略和dropout技术进一步防止模型过度拟合。
[0069]
(4)采用类重新加权的方法,增加阳性样本的影响,迫使模型学习占少数的阳性样本的抽象机制。
[0070]
(5)本发明中,深度学习模型是基于keras 2.1.6和tensorflow 1.13.1实现的,模型训练和测试在具有ubuntu 18.04.1lts系统并配备gpu nvidia tesla v100-pcie-32gb的工作站上进行。
[0071]
其中,通过十折交叉验证、独立测试、模型泛化能力测试和对未知赖氨酸乙酰化位
点的识别能力四种类型的实验来评估提出的模型,包括步骤:
[0072]
(1)采用十折交叉验证在相同基准训练数据集下比较基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型和其他预测方法的性能;
[0073]
(2)采用独立测试的方式,进一步比较基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型与其他模型的预测能力;
[0074]
(3)采用泛化实验的方式进一步验证基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型具有较好的泛化能力;
[0075]
(4)在独立测试集上,验证排名前20的候选位点,评估基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型识别未知的赖氨酸乙酰化位点的能力。
[0076]
区别于现有技术,本发明所述的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法,引入蛋白质结构特性,将其与蛋白质原始序列、氨基酸理化属性相结合以构建位点特征空间;采用模块化密集卷积网络捕获不同层级的特征信息,在特征学习过程中减少信息丢失和信息串扰;并引入压缩-激发层来评估不同特征的重要性,提高网络的抽象能力,以识别潜在的赖氨酸乙酰化位点。本发明能够有效地解决现有方法仅考虑蛋白质序列层面信息和特征学习效率低下的问题,更准确的预测了潜在的赖氨酸乙酰化位点,降低赖氨酸乙酰化位点的验证成本,提高了赖氨酸乙酰化修饰的研究效率。
附图说明
[0077]
图1是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法的流程示意图;
[0078]
图2是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,所收集的人类数据集信息,其中cd-hit的阈值为0.4;
[0079]
图3是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,密集卷积网络的示意图;
[0080]
图4是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,压缩-激发模块的示意图;
[0081]
图5是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,0.4去冗余阈值下,人类训练数据集上不同方法的十倍交叉验证性能,粗体为相应指标下最高值;
[0082]
图6是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,不同预测方法在0.4去冗余阈值下,人类独立测试数据集上的预测性能;
[0083]
图7是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,各模型在0.4去冗余阈值下,大肠杆菌独立测试数据集上预测性能,粗体为相应指标下最高值;
[0084]
图8是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法中,0.4去冗余阈值下,人类独立测试乙酰化蛋白质的前20个候选位点的预测结果,粗体是实际发生乙酰化修饰的位点。
具体实施方式
[0085]
下面结合具体实施方式对本发明的技术方案作进一步更详细的描述。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动的前提下所获得的所有其他实施例,都应属于本发明保护的范围。
[0086]
参阅图1,图1是本发明提供的一种基于模块化密集卷积网络的赖氨酸乙酰化位点预测方法的流程示意图。
[0087]
该方法的步骤包括:
[0088]
s110:从蛋白质结构特性、蛋白质原始序列和氨基酸理化属性信息三个方面描述赖氨酸乙酰化位点,构建位点初始特征空间。
[0089]
所述步骤s110包括:
[0090]
1、赖氨酸乙酰化位点的实验数据收集和预处理;
[0091]
2、通过编码方式将收集到的蛋白质数据转化为数值向量,构建位点初始特征空间,并作为预测模型的输入。
[0092]
赖氨酸乙酰化位点的实验数据收集和预处理,包括步骤:
[0093]
从蛋白质赖氨酸修饰数据库(plmd)收集并下载了6078条、3645条和1860条经实验验证的人类、小家鼠和大肠杆菌赖氨酸乙酰化蛋白质数据。
[0094]
考虑到spider3服务器无法处理含有非标准氨基酸的蛋白质序列,本发明手动删除了这些蛋白质序列。以人类这一物种为例,利用cd-hit工具进行序列去冗余避免序列同源性较大而造成模型的偏差,阈值设定为0.4,保留了4977条乙酰化蛋白质序列。本发明将过滤后的4977条乙酰化蛋白质序列随机选择10%(498条)构建独立测试数据集,剩余乙酰化蛋白质序列作为训练数据集,便于与其他赖氨酸乙酰化位点预测器进行比较。
[0095]
通过编码方式将收集到的蛋白质数据转化为数值向量,构建位点初始特征空间,并作为预测模型的输入,包括步骤:
[0096]
(1)使用one-of-21编码位点的蛋白质原始序列信息,对于长度为l的基序,将得到l
×
21维的蛋白质原始序列信息的向量表示;
[0097]
(2)采用atchley因子编码位点的氨基酸理化属性信息,每个氨基酸残基由5个atchley因子表示,对于长度为l的基序,将得到l
×
5维的氨基酸理化属性信息的向量表示;
[0098]
(3)通过spider3获取蛋白质结构特性信息,包括3种属性中的8个指数,即二级结构:α螺旋p(h)、β链p(c)、γ环p(e),局部骨干扭转角:ψ、θ、τ,可及表面积:asa。对于长度为l的基序,将得到l
×
8维的蛋白质结构特性信息的向量表示。
[0099]
s120:采用模块化密集卷积网络,从位点的初始特征空间分别提取蛋白质结构特性、蛋白质原始序列和氨基酸理化属性的高级特征,通过密集跳跃连接同时关注低层级特征和高层级特征。
[0100]
所属步骤s120包括:
[0101]
1、引入模块化网络结构的设计思想,构建结构、序列和理化三个信息模块;
[0102]
2、采用堆叠密集卷积块对每个模块进行高级特征的提取,通过密集跳跃连接同时考虑低层级和高层级特征,实现不同层级特征间的信息互补。
[0103]
引入模块化网络结构的设计思想,构建结构、序列和理化三个信息模块,包括步
骤:
[0104]
基于蛋白质结构特性、蛋白质原始序列和氨基酸理化属性分别构建了结构模块、序列模块和理化模块和三个特征提取子模块,各子模块间参数空间相互独立,有效避免了三类信息之间的串扰,提高特征的质量。
[0105]
采用堆叠密集卷积块对每个模块进行高级特征的提取,通过密集跳跃连接同时考虑低层级和高层级特征,实现不同层级特征间的信息互补,包括步骤:
[0106]
由于结构模块、序列模块和理化模块网络结构相同,这里仅对序列模块进行说明:
[0107]
(1)首先,序列模块接收长度为l的位点基序的one-of-21编码作为输入,然后通过一维卷积层生成蛋白质原始序列信息的低级特征图,如公式(1)所示。
[0108]
x0=σ(i*w+b)
ꢀꢀꢀꢀꢀꢀꢀꢀ
(1)
[0109]
其中,i为one-of-21编码向量。为权重矩阵,s为过滤器的大小(s=3),d是过滤器数量(d=96)。b为偏置项,σ为激活函数。x0为一维卷积层的输出,大小为l
×
d。
[0110]
(2)采用密集卷积块提取蛋白质原始序列信息的高级特征表示,密集卷积过程如公式(2)所示。
[0111]
x
l
=σ([x0;x1;...;x
l-1
]*w
′
+b
′
)
ꢀꢀꢀꢀꢀꢀ
(2)
[0112]
其中,x
l-1
为密集卷积块中第l-1个卷积层生成的特征图,[
·
]表示沿特征维串联。为权重矩阵,d
′
为密集卷积块中1到l-1层卷积的过滤器总数,d
″
为密集卷积块中第l个卷积层过滤器数(d
″
=32)。b
′
为偏置项,σ为激活函数,x
l
表示密集卷积块中第l个卷积层生成的特征图。密集卷积块的输出为低级特征图x0与密集卷积块中每个卷积层生成的特征图x1,x2,...,x
l
的特征维串联,即[x0;x1;...;x
l
]。
[0113]
(3)采用过渡层将(2)得到的蛋白质原始序列信息的特征图再进行卷积运算和激活操作,过渡层过程如公式(3)所示。
[0114]
x=σ([x0;x1;...;x
l
]*w
″
+b
″
)
ꢀꢀꢀꢀꢀꢀ
(3)
[0115]
其中,为权重矩阵,s
′
为过滤器的大小(s
′
=1)。b
″
为偏置项,σ为激活函数,x为过渡层的输出。之后,对过渡层结果进行平均池化操作,以减小特征图维数,降低模型过拟合风险。
[0116]
(4)重复(2)、(3)步骤,构成堆叠密集卷积块。第四次的(2)步骤后不进行(3)步骤,而是使用全局平均池化替代。
[0117]
经过上述过程,序列模块提取到位点的蛋白质原始序列的高级特征x
(seq)
。
[0118]
类似,理化模块和结构模块也通过上述过程提取到位点的氨基酸理化性质和蛋白质结构特性的高级特征。
[0119]
s130:引入压缩-激发(se)层评估特征的重要性,加权每个特征图,实现三类信息的自适应动态融。
[0120]
所属步骤s130包括:
[0121]
1、引入压缩-激发(se)层评估特征的重要性,加权每个特征图;
[0122]
2、蛋白质结构特性、蛋白质原始序列和氨基酸理化属性三类信息的自适应动态融合。
[0123]
引入压缩-激发(se)层评估特征的重要性,加权每个特征图,包括步骤:
[0124]
以序列模块为例进行说明:
[0125]
(1)压缩(squeeze):采用全局平均池化将序列模块提取到的高级特征x
(seq)
的全局空间信息压缩到通道描述符中,压缩过程如公式(4)所示。
[0126][0127]
其中,z
c
表示x
(seq)
的第c个特征图的通道统计信息,f
sq
(
·
)表示压缩操作,w和h分别表示特征图的宽和高。将x
(seq)
的每个特征图都计算统计信息后,得到x
(seq)
的通道描述符
[0128]
(2)激发(excitation):采用两个全连接层(fc)捕获x
(seq)
的通道依赖性,学习x
(seq)
中每个特征图的特异性权值,激发过程如公式(5)所示。
[0129]
s=f
ex
(z,w)=σ(w2*δ(w1*z))
ꢀꢀꢀꢀ
(5)
[0130]
其中,表示激发操作后学习到的x
(seq)
中每个特征图的特异性权值,f
ex
(
·
)表示激发操作。δ和σ分别表示两个全连接层的激活函数,前者为relu函数,此层为具有参数的维度约减层,其缩减比为r(r=16);后者为sigmoid函数,此层为具有参数的维度还原层,确保s的维度与特征x
(seq)
的通道数相同。
[0131]
(3)特征缩放(scale):通过以下激活缩放x
(seq)
得到se层的输出其中为的任意一个元素,的计算如公式(6)所示。
[0132][0133]
其中,表示将特征图上的每个值与权值s
c
相乘。
[0134]
类似,理化模块和结构模块也通过se层得到加权后的高级特征。
[0135]
蛋白质结构特性、蛋白质原始序列和氨基酸理化属性三类信息的自适应动态融合,包括步骤:
[0136]
se层是基于全局平均池化和两个全连接层(fc)实现的,且结构模块、序列模块和理化模块的网络结构相同,都通过se层得到加权后的高级特征。然后,串联每个子模块的输出,得到用于分类的融合特征由于se层为不同的特征图进行加权,使得特征融合过程具有自适应动态特性。
[0137]
s140:基于融合特征和softmax层构建赖氨酸乙酰化位点分类器,预测潜在的赖氨酸乙酰化位点。
[0138]
所属步骤s140包括:
[0139]
softmax层接收高级特征作为输入,经过加权求和和激活操作后得到样本的预测类别,softmax层的前向传播过程如公式(7)所示。
[0140][0141]
其中,为权重矩阵,为偏置项。p(y=i|x)表示样本x预测为i∈{0,1}类的概率,概率最大所对应的类别即为softmax分类器的预测类别。
[0142]
s150:训练基于模块化密集卷积网络赖氨酸乙酰化位点预测模型。
[0143]
所属步骤s150包括:
[0144]
1、采用交叉熵作为代价函数,以最小化训练误差:
[0145][0146]
其中,n为训练样本总数,y
j
为第j个输入基序的真实标签,x
j
为第j个输入基序。
[0147]
2、在训练中采用l2正则化,以减轻过拟合的影响,模型的最终目标函数为:
[0148]
min
w
(l
c
+λ∑(||w||2)2)
ꢀꢀꢀꢀꢀꢀꢀ
(9)
[0149]
其中,λ为正则化系数,||w||2为权重矩阵的l2范数。
[0150]
3、采用adam优化器对目标函数进行优化,学习率和批处理分别设置为0.0001和1000。采用early stopping策略和dropout技术进一步防止模型过度拟合。
[0151]
4、采用类重新加权的方法,增加阳性样本的影响,迫使模型学习占少数的阳性样本的抽象机制。
[0152]
5、本发明中,深度学习模型是基于keras 2.1.6和tensorflow 1.13.1实现的,模型训练和测试在具有ubuntu 18.04.1lts系统并配备gpu nvidia tesla v100-pcie-32gb的工作站上进行。
[0153]
s160:通过十折交叉验证、独立测试、模型泛化能力测试和对未知赖氨酸乙酰化位点的识别能力四种类型的实验来评估提出的模型。
[0154]
所属步骤s160包括:
[0155]
1、采用十折交叉验证在相同基准训练数据集下比较基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型和其他预测方法的性能;
[0156]
2、采用独立测试的方式,进一步比较基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型与其他模型的预测能力;
[0157]
3、采用泛化实验的方式进一步验证基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型具有较好的泛化能力;
[0158]
4、在独立测试集上,验证排名前20的候选位点,评估基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型识别未知的赖氨酸乙酰化位点的能力。
[0159]
其中,采用十折交叉验证在相同基准训练数据集下比较基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型和其他预测方法的性能,包括步骤:
[0160]
(1)采用了十折交叉验证的方式,将本发明的模型与其他现有的赖氨酸乙酰化位点预测模型:musitedeep、capsnet、deepacet、pskacepred、ensemblepail、gps-pail2.0和proacepred进行比较。
[0161]
(2)采用六种统计度量指标评估模型的性能,包括灵敏度(sn)、特异性(sp)、准确度(acc)、精确率(pre)、马修相关系数(mcc)和几何均值(g-mean),它们的定义如下:
[0162][0163][0164][0165][0166][0167][0168]
其中,tp、tn、fp和fn分别为真阳性、真阴性、假阳性和假阴性。当阳性样本和阴性样本不平衡时,mcc和g-mean指标可以很好地反映模型质量。此外,我们还采用接收器工作特性(roc)曲线下面积(auc)和精确率召回率(pr)曲线下面积(aupr)来衡量模型整体性能,auc和aupr值越高,表明模型整体表现越好。模型的比较结果见说明书附图。
[0169]
其中,采用独立测试的方式,进一步比较基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型与其他方法的预测能力,包括步骤:
[0170]
对于具有独立工具的模型,采用训练数据训练模型,然后在独立测试数据集上进行潜在的赖氨酸乙酰化位点预测,对于提供web服务的模型,仅基于独立测试数据集测试其预测性能。结果表明基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型具有最高的mcc、g-mean、auc和aupr,在独立测试数据集上表现最优,相较于其他预测方法具有更好的赖氨酸乙酰化位点预测能力。独立测试的结果见说明书附图。
[0171]
其中,采用泛化实验的方式进一步验证基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型具有较好的泛化能力,包括步骤:
[0172]
采用了泛化实验的方式,在人类数据集0.3去冗余阈值下、小家鼠数据集0.4和0.3去冗余阈值下以及大肠杆菌数据集0.4和0.3去冗余阈值下对赖氨酸乙酰化位点进行预测。由结果得出基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型具有较好的泛化能力,能够适用于不同物种数据集,这为更多的其他物种的赖氨酸乙酰化修饰位点的预测提供了可用参考。泛化能力测试的结果见说明书附图。
[0173]
其中,在独立测试集上,验证排名前20的候选位点,评估基于模块化密集卷积网络的赖氨酸乙酰化位点预测模型识别未知的赖氨酸乙酰化位点的能力,包括步骤:
[0174]
根据独立测试集的结果列出了本发明的模型预测为赖氨酸乙酰化的前20个候选位点,并在在赖氨酸修饰数据库plmd和蛋白质数据库uniprot(https://www.uniprot.org)中手动查验这20个候选位点。通过统计验证结果,发现20个候选位点中有13个是真正乙酰化的,占比为65%。人类独立测试乙酰化蛋白质的前20个候选位点结果见说明书附图。
[0175]
以上仅为本发明的实施方式,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等效结构或等效流程变换,或直接或间接运用在其他相关的技术领域,均同理包括在本发明的专利保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1